





Abstract
Aims Familial dilated cardiomyopathy (FDCM) is associated with mutations in more than 10 genes, but genes mutation frequencies and associated clinical features remain largely unknown. Here, we performed a mutation analysis of four genes involved in FDCM in a population of idiopathic DCM.
Methods and results A SSCP and sequencing mutation screening of all the exons coding for beta myosin heavy chain (MYH7 gene), cardiac T troponin (TNNT2 gene), phospholamban (PLN gene), and the cardio-specific exon of metavinculin (VCL gene) were performed in 96 independent patients (54 familial and 42 sporadic). It led to the identification of eight heterozygous mutations, seven new ones in MYH7, and the already described R141W mutation in TNNT2. MYH7 mutations (in five familial and two sporadic cases) substitute residues located either in the head (I201T, T412N, A550V) or tail domains (T1019N, R1193S, E1426K, R1634S) of the protein. DCM was not associated with skeletal myopathy or conduction defects in any patients. Contrasting clinical features were observed between MYH7 and TNNT2 mutations carriers. In MYH7 vs. TNNT2, mean age at diagnosis was late (P<0.03), penetrance was incomplete in adults (56 vs. 100%), and mean age at major cardiac event was higher (P<0.04).
Conclusion We have identified seven mutations in MYH7, one in TNNT2, and none in PLN or in the VCL cardio-specific exon. MYH7 appears as the most frequently mutated gene in our FDCM population (∼10%), and mutation carriers present with delayed onset, in contrast to TNNT2.
for the editorial comment on this article (doi:10.1093/eurheartj/ehi208)
Introduction
Idiopathic dilated cardiomyopathy (DCM) is a major cause of heart failure, sudden death, and heart transplantation. The phenotype can be characterized by an isolated cardiac dysfunction (isolated DCM) or include conduction defects (atrioventricular block or sinus node dysfunction) and/or skeletal muscular disorders. The majority of patients present a sporadic DCM (SDCM), however, a familial transmission (FDCM) is observed in 25–35% of the cases.1–3
Mutation analysis on cohorts of patients with autosomal dominant inheritance led to the identification of null and missense heterozygous mutations in 12 different genes encoding sarcomeric, cytoskeletal, or regulatory proteins associated with isolated FDCM: the α-cardiac actin (ACTC), desmin (DES), δ-sarcoglycan (SCGD), vinculin (VCL), titin (TTN), T troponin (TNNT2), α-tropomyosin 1 (TPM1), muscle LIM protein (MLP), phospholamban (PLN), ZASP (LDB3), and β-myosin heavy chain (MYH7) genes.4–11 Moreover, mutations in the lamin (LMNA) or the two X-linked dystrophin (DMD) and taffazin (TAZ) genes are responsible for FDCM frequently associated with specific phenotypes including conduction defects and/or muscular skeletal dystrophy.4,12 Finally, up to seven genetic loci have also been identified but without subsequent disease gene characterization.4
We have previously reported the mutation screening of several known disease genes in a French population composed of independent SDCM and FDCM cases. These screened genes included ACTC, DES,13 and SCGD,14 and we confirmed their low mutation rate, whereas LMNA15 mutations were strongly associated with DCM cases presenting with additional phenotypes. In order to better estimate the incidence of mutations in DCM and associated phenotypes in this population, we extended our mutation analysis to the coding sequences of four more disease genes implicated in DCM: MYH7 (OMIM 160760), TNNT2 (OMIM 191045), PLN (OMIM 172405), and the cardio-specific exon of VCL (OMIM 193065), which is included in the transcript encoding metavinculin in heart, a cytoskeletal protein implicated in anchoring of the F-actin to plasma membrane.
Methods
Clinical evaluation
Ascertainment of subjects recruited in the study was based on physical examination including muscular testing, ECG, coronary angiography, and 2D-echocardiography. Holter-ECG and serum creatine kinase level were obtained when possible. The diagnosis of DCM was established in accordance with the criteria used by a European consortium described elsewhere.16 Patients were classified as familial cases when at least two first-degree patients in the same family were affected. All first-degree relatives of patients were proposed to be included in the study and ∼80% of them gave consent. Cases were considered sporadic when no evidence of familial disease was observed or when no relatives could be clinically evaluated. Relatives in families were classified as unknown in the presence of minor non-significant abnormality or because of interaction with another cardiac disease (such as coronary artery disease). Written informed consent for all subjects (index cases and relatives) was obtained in accordance with the study protocol approved by the local ethics committee. Inheritance in familial forms was reviewed and families with X-linked or mitochondrial inheritance were excluded. A total of 96 consecutive unrelated index cases were enrolled and investigated, including 54 FDCM and 42 SDCM cases. No index patient declined to take part in the study. The characteristics of the studied population have been described previously.14 Patients were mainly of European origin (94%) and recruited in France. The male/female gender ratio was 63:36 and mean age was 47±18 years. Most cases were presenting with isolated DCM (n=92; 96%), some were characterized by DCM and skeletal muscular dystrophy or conduction defect (n=3; 3%), or early atrial fibrillation (n=1; 1%).
Mutation analysis
Blood samples were collected and DNA was extracted from peripheral lymphocytes using a standard protocol at the Généthon Bank (Evry, France). Oligonucleotides were designed in intronic sequences flanking coding exons according to the following accession number sequences: MYH7, AJ238393; TNNT2, X74819; PLN, NM_002667; and VCL, NM_014000. Amplifications were conducted following standard PCR protocol.14 Specific conditions and oligonucleotide sequences for each PCR amplification are fully available on request. For MYH7 and PLN, each amplified DNA fragment was analysed by SSCP (single strand conformation polymorphism) using two different temperature conditions as previously described.14 Abnormal profiles observed after silver staining were sequenced on an ABI Prism 3100 Genetic Analyser (Applied Biosystems). The two other genes (TNNT2, VCL) were directly sequenced after exon specific PCR amplifications. Identified mutations were confirmed by sequencing on an independent PCR product as well as the segregation of the mutation with the disease in each family.
Genotyping of identified molecular variants
Identified molecular variants were considered as potential mutations when the encoded protein sequence was changed. Synonymous variants were considered as polymorphisms. No variants were identified in the intron–exon junctions.
All suspected mutations were genotyped by PCR–RFLP (restriction fragment length polymorphism) or original SSCP in all family members available and in a control population consisting of 236 ethnically matched subjects from the MONICA (Monitoring in Cardiovascular Disease) study and aged 18–64 with no identified cardiovascular pathology. Table 1 presents the genotyping conditions used for each of the new non-synonymous molecular variants identified. Polymorphism frequencies (Table 2) were estimated by counting from screening results of the 96 index cases (sequence traces or SSCP patterns).
Statistical analyses
Values were expressed as mean±SD. Continuous variables were compared between two groups with a two-sided Mann–Whitney test. A value of P<0.05 was considered as significant.
Results
In order to estimate the sensitivity of our SSCP protocol, we extensively double-strand sequenced 11 exons of the human MYH7 gene on more than 400 subjects and observed five polymorphisms presenting a rare allele frequency of >1%. All five polymorphisms were detected by our SSCP protocol, setting the sensitivity to 100% in that limited assay compared with double strand sequencing. It suggests, therefore, a very high sensitivity of our SSCP mutation detection protocol.
Polymorphisms
We identified 23 exonic polymorphisms (Table 2). All were synonymous single base substitutions except the known S1491C in MYH717 and one rare non-synonymous polymorphism (P938A) observed in one subject and one healthy control in VCL exon 19.
Mutations
No mutation was identified in the coding exon of PLN gene or in the cardiac specific exon of VCL. Conversely, the MYH7 coding sequence mutation screening led to the identification of seven new heterozygous missense mutations due to single nucleotide substitutions (Figure 1). Five were found in familial forms and two in sporadic cases. The MYH7 gene was therefore involved in 9% of all FDCM (5/54), 10% of families with isolated common FDCM (5/50), and 5% of SDCM (2/42). One familial and one sporadic cases carried the same and already identified R141W substitution in TNNT2, raising the frequency of this single mutation to 2% (Figure 1). None of these mutations was identified in at least 400 control alleles, strongly suggesting that they are not neutral polymorphisms. Molecular alignment shows conservation of TNNT2 (data not shown) and MYH7 (Figure 2) mutated residues among species and isoforms.
Familial mutations
In family A, all three affected members were carriers of a T688 C substitution (exon 7) which is predicted to lead to an I201T mutation. One family member (II-1) was a carrier of the mutation and had isolated left ventricular dilatation but without systolic dysfunction. In family B, a C1735T substitution (exon 16) leading to an A550V mutation was present in two affected members of the family. In family C, we identified a C3142A substitution (exon 24) substituting a threonine to an asparagine (T1019N). There were two affected carriers, including an obligate one, in this family. The mutation was also found in three subjects with unknown clinical status, including two patients with left ventricle dilatation and systolic dysfunction but with significant coronary artery disease.
There were also four healthy carriers in the second generation (age ≤48 years), strongly suggesting a late onset of the disease caused by this mutation. In family D, we identified a C3663A substitution (exon 27) driving the replacement of the basic amino acid arginine into a neutral polar serine residue (R1193S). The family presented 10 mutation carriers including an obligate one. As in family C, penetrance was incomplete with four affected carriers, three individuals with uncertain status, and three healthy. Finally, in family E, a G4362A (exon 31) substitution was identified giving rise to a glutamic acid to lysine replacement (E1426K). There were only two known carriers in this family and both were affected. Moreover, patient II-1 was also affected and, despite the lack of available DNA, was an obligate carrier of the mutation.
Sporadic mutations
We found a C1321A substitution (exon 13) and a C4986T substitution (exon 34) leading to the mutation T412N and R1634C, respectively. It could not be stated whether they were de novo mutations as parents were deceased.
There were five affected members in family F including four carriers of the TNNT2 R141W mutation. We also identified the same R141W mutation in sporadic case C, which is very likely a de novo mutation as it was absent from both healthy parents. However, the possibility of false paternity could not be ruled out.
Clinical features
Detailed phenotypic data on the heterozygous subjects with a mutation are reported in Table 3 and pedigrees are presented in Figure 1. All subjects were of European origin except sporadic C who originates from Magrib. All patients displayed isolated forms of DCM, except one relative with AV block II in family D. In family C, one of the relatives who had an unknown clinical status (subject II-4) did not carry the mutation. She had significant aortic stenosis, symmetrical LV hypertrophy, normal LV diameter, and mildly depressed ejection fraction.
In MYH7 mutation carriers, mean age at diagnosis was 48±17 years. Only 18 among 30 mutation carriers had symptoms, and onset of symptoms occurred after 30 years of age in all cases except one. Penetrance of mutations was incomplete in familial forms (P=14/25 or 56% in adults). Among 12 mutation carriers without DCM, four had isolated and mild ventricular dilatation and one had isolated and mild depressed systolic function. Prognosis, as assessed by the follow-up of mutation carriers and by family history, was characterized by three cardiac deaths (two mutation carriers and one without available DNA) and three heart transplantations (two mutation carriers and one without available DNA). Mean age of these major cardiac events was 55±12 years. Survival rate at 40 and 65 years in DCM families was 96 and 65%, respectively.
In family C, the myocardial biopsy of an affected subject (II-3) revealed no cellular disorganization but extensive fibrosis (data not shown). There was no myosin or desmin abnormal immunolabelling pattern, suggesting a normal sarcomeric organization.
All TNNT2 mutation carriers (five subjects) were symptomatic. Penetrance was complete in the familial form but the size of the family is small. DCM occurred very early in two patients (<3 months of age) and one of them died within few days. There were three cardiac deaths (one in a mutation carrier and two without available DNA in the family), and mean age was 21±24 years.
Comparisons between gene-specific mutation carriers
When comparing the mutation carriers, mean age at diagnosis was lower in the TNNT2 group than in the MYH7 group (22.6±21 vs. 48.2±17 years, P=0.029) and mean age at major cardiac events was significantly lower (21.3±24 vs. 55.5±12 years, P=0.039). When age at diagnosis was compared in index cases only, mean age was 0.2±0 (n=2, TNNT2) vs. 41.6±12.6 years (n=7, MYH7), P=0.04.
Discussion
The prevalence and clinical features of DCM related to four genes, previously reported as disease genes were investigated in 96 subjects with DCM. We have found seven new mutations in MYH7 in seven index cases, one mutation in TNNT2 in two index cases, and no mutations in PLN or exon 19 of VCL.
Vinculin and phospholamban
The mutation frequency in PLN and VCL cardio-specific exon 19 appears to be very low in the population screened in the present study (no mutation). Estimates from previous reports evaluate the frequency of FDCM causing mutations in PLN (one mutation) and VCL exon 19 (two mutations) to 5 and 3%, respectively.6,8 Our results are indicative of a smaller frequency (<2% in FDCM), confirming that mutations in both genes are rare in FDCM.
Cardiac T troponin
We identified one mutation in two index cases in TNNT2. Previous screening on TNNT2 in FDCM has identified only two different mutations (ΔLys210 and R141W).10,18 To date, the more frequent of these two mutations (ΔLys210) has been found four times at frequencies ranging from 10 to <2% depending on the studies.10,11,19 Here, we did not observe the ΔLys210 mutation, but R141W with a 2% frequency. Interestingly, in our screening, R141W was observed twice. In addition to a familial form, the mutation is observed in a sporadic case from a different ethnic origin as a likely de novo mutation, suggesting independent events rather than a founder effect at this position. In accordance with previously described phenotypes associated with ΔLys210 and R141W, the clinical feature observed in our population was characterized by early onset and severe form of FDCM.
These findings may have implications, namely, for the purpose of molecular diagnosis strategy specifically designed to identify ΔLys210 and R141W, which seem to be involved in 2% of FDCM cases and more likely associated with early onset.
Beta myosin heavy chain
We identified seven new mutations responsible for FDCM in MYH7. All identified mutations affect amino acids with various but high degrees of conservation among cardiac myosin isoforms from different species, suggesting the functional importance of these residues (Figure 2). Moreover, none of the mutations was present in more than 400 chromosomes from a control population. Together with the very low occurrence of myosin heavy chain mutation or polymorphism in the general population, as observed by systematic MYH7 screening20 and an almost complete association of non-conservative MYH7 mutations with cardiac diseases such as hypertrophic cardiomyopathy (HCM) or DCM, these genetically based observations strongly support a role in disease for the seven newly identified mutations in our study.
In our population, MYH7 appears so far to be the most prevalent gene involved in ∼10% of FDCM and extends previous results by other groups to a broader population.10 This is supported by additional screening of other genes conducted in at least 80% of the families screened in the present study. Accordingly, in addition to the three other genes less prevalent than MYH7 screened here (TNNT2, VCL, PLN), we previously reported the absence of mutations in the SCGD, ACTC, and DES genes.13,14 In that previous screening of the same population, we also identified three families with LMNA mutations, all presenting non-isolated FDCM, i.e. associated with myopathy, conduction defects, or arrhythmia. Thus, the prevalence of LMNA mutation in isolated FDCM is null, and it reaches 50% of DCM families with non-isolated forms on the basis of our experience.15 Other publications reported identification of the LMNA gene mutations responsible for FDCM.21,22 It is of note that almost all the mutations identified have been found in subjects or families presenting with non-isolated forms of DCM. The exact frequency of LMNA mutation in the whole FDCM population is difficult to assess given the phenotypic heterogeneity in families included in the different published studies. However, LMNA appears to be only marginally implicated in common forms with isolated FDCM. As other disease genes are observed only in a few families, (<5%), MYH7 appears so far as the most prevalent gene reported in common and familial forms of DCM.
The phenotype in MYH7 mutation carriers offers some peculiarity. Mean age at diagnosis was late, penetrance was incomplete, onset of symptoms was delayed, and mean age of cardiac death or heart transplantation was 55±12 years. Interestingly, age at diagnosis and age at major cardiac event were also significantly higher than those observed in patients with DCM and a mutation in TNNT2 (although there was a wide distribution). The MYH7 gene appears, therefore, associated with a late-onset DCM in our population. Our findings are in contrast with previous descriptions by Kamisago et al.10 and Daehmlow et al.11 of such patients in whom early onset was reported. Several reasons could explain this apparent discrepancy. First, the structure of the studied populations is different: our findings are based on seven independent mutations in 30 mutation carriers, whereas Kamisago et al. explored two large kindreds. Secondly, Daehmlow et al.11 have selected DCM cases based only on early onset disease and did not analyse relatives, blunting any attempt to evaluate penetrance or to analyse genotype–phenotype correlation. Finally, it is likely, as observed in hypertrophic cardiomyopathy caused by MYH7 mutations, that various mutations in a same gene give rise to contrasted phenotypes, possibly because of the specific effect of the mutation itself, of environmental, or of modifying gene effects.
In the present study, we report the mutation screening of all the coding sequence of the MYH7 gene and not only exons encoding the head motor domain of the protein. The identification of four mutations in the tail domain emphasizes the importance of a complete MYH7 gene screening as recently suggested in HCM.17,23
Several findings indicate that families and patients described in the present work have primary DCM and not infrequent end-stage HCM with systolic dysfunction. HCM was never observed in the present families, even at the beginning of the cardiac disease. No sign of echocardiographic or electrocardiographic hypertrophy was observed during inquest in patients or relatives [mean interventricular septum (IVS) thickness was 8.3±1.5 mm], and there was no myocardial disarray in the available myocardial biopsy.
Though functional studies are needed to understand the pathophysiological mechanisms induced by mutations, the absence of a clear localization of the various mutations in a discrete functional domain of the protein suggests that different mechanisms could be involved in the disease process. Three of the identified mutations are located in the head motor domain involved in actin–myosin interaction or ATP/ADP processing. Numerous mutations affecting this domain of the protein have been associated with HCM as well as DCM. Conversely, the other four mutations take place in the rod domain of the Myosin heavy chain molecule. The four mutated residues observed here occurred at conserved positions in the rod domain and three of them give rise to a change in the amino acid net charge. This suggests that rod mutations could have an adverse effect by impairing myosin rod structure and assembly.
In HCM patients, more than 80 different disease mutations have been identified to date in MYH7.23 Interestingly, none of the DCM mutations identified in the present study has been previously implicated in HCM, suggesting mutation-specific mechanisms. Recent systematic candidate gene screening in DCM indicates an initially unexpected large overlap between HCM and DCM responsible genes. A future challenge will be, therefore, to identify the precise mechanisms leading from mutations to specific disease.
In conclusion, our findings confirm the genetic heterogeneity of FDCM with a low occurrence of PLN and VCL mutation, a 2% frequency of TNNT2 mutation (R141W is associated with high penetrance and early onset), and a prominent role of MYH7 in DCM with a 10% mutation frequency in familial forms and delayed onset of the disease in the studied population.
Acknowledgements
These studies would not have been possible without the invaluable assistance of patients and family members. We thank Dr Renard, Professor M. Goenen, and Professor P. Gibelin for their clinical collaboration. We also thank Dr S. Tezenas du Montcel for statistical reviewing of the manuscript. This work is dedicated to the memory of Jean Leducq and would not have been possible without a generous grant from the Leducq Foundation and the ‘Fondation de France’
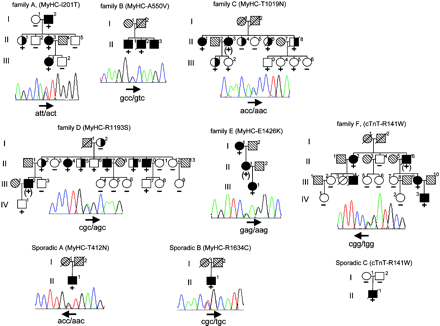
Figure 1 Pedigree drawings of DCM families carrying mutations and sequence electrophoregram representative of each heterozygous mutation. Pedigree symbols: +, heterozygous carrier of mutation; (+), obligate mutation carrier; −, no mutation; filled symbols, DCM affected; open symbol, unaffected subject; half-filled symbol, unknown status; slashed symbol, deceased individuals; hatched symbols, no phenotypical data available. Arrows underlined mutated codon sequences and codon reading orientation.
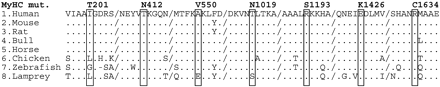
Figure 2 Multiple alignment of myosin heavy chain (MyHC) was performed with the ClustalW program using the default parameters. Mutated residues are indicated in the top sequences (MyHC mut) with amino acid position related to the human protein. Dots are for residues identical to the human protein. Accession numbers of aligned protein sequences are: (i) gi4507627; (ii) gi6981666; (iii) gi1717770; (iv) gi27807485; (v) gi136399; (vi) gi33870023; (vii) gi6755841; (viii) gi33465568.
Genotyping conditions for identified mutations and non-synonymous molecular variants
| Molecular variant | Method | Number of controls genotyped | Restriction enzyme; SSCP conditions | PCR primer sequence (5′→3′) | Size (bp) | Allele sizes (bp) | PCR conditions (Temp/[MgCl2]) | |
|---|---|---|---|---|---|---|---|---|
| MYH7 gene | ||||||||
| I201T | SSCP | 224 | 10°C/1h | F: CACTGCCCAATAAGCCCC | 165 | na | na | 65°C/2 mM |
| (mut.) | R: CAGGACCTTGGAGGGCAG | |||||||
| T412N | RFLP | 232 | StyI | F: TTACAGGCATGAACCACACACC | 266 | 169+97 | 266 | 60°C/1 mM |
| (mut.) | R: GTGAACTTGAAAACTCTCATCCC | |||||||
| A550 V | RFLP | 206 | HaeIII | F: GAGCAGAATCCATGTCACCT | 266 | 135+ | 157+ | 60°C/1 mM |
| (mut.) | R: TGCAGCCAGCCAATGATGTT | 109+22 | 109 | |||||
| T1019N | SSCP | 231 | 25°C/2h | F: GACCATACTGACCTTGACCC | 353 | na | na | 60°C/1 mM |
| (mut.) | R: AGACATGGCATATCTAGGCC | |||||||
| R1193S | RFLP | 236 | HhaI | F: TGCAGATCGAGATGAACAAG | 295 | 193+80 | 273+22 | 60°C/1 mM |
| (mut.) | R: GTGGGATTAGGAAGTTGGA | +22 | ||||||
| E1426K | RFLP | 236 | TaqI | F: ATCCTCCCCACCCTCTGC | 299 | 140+86 | 213+86 | 60°C/1 mM |
| (mut.) | R: GAGGATGGCTCTGGCCTCT | +73 | ||||||
| R1634C | RFLP | 234 | PvuIIa | F: AGCTCAGCCACGCCAGC | 135 | 135 | 117+18 | 60°C/1 mM |
| (mut.) | R: CCTGTATCAAGACACTACTGCTTACG | |||||||
| VCL gene | ||||||||
| P943A | RFLP | na | BslI | F: TCCTAGGCAGGCTTTGGTTA | 237 | 189+48 | 237 | 60°C/2 mM |
| (polym.) | R: TTGGATGGCATTAACAGCAG | |||||||
| Molecular variant | Method | Number of controls genotyped | Restriction enzyme; SSCP conditions | PCR primer sequence (5′→3′) | Size (bp) | Allele sizes (bp) | PCR conditions (Temp/[MgCl2]) | |
|---|---|---|---|---|---|---|---|---|
| MYH7 gene | ||||||||
| I201T | SSCP | 224 | 10°C/1h | F: CACTGCCCAATAAGCCCC | 165 | na | na | 65°C/2 mM |
| (mut.) | R: CAGGACCTTGGAGGGCAG | |||||||
| T412N | RFLP | 232 | StyI | F: TTACAGGCATGAACCACACACC | 266 | 169+97 | 266 | 60°C/1 mM |
| (mut.) | R: GTGAACTTGAAAACTCTCATCCC | |||||||
| A550 V | RFLP | 206 | HaeIII | F: GAGCAGAATCCATGTCACCT | 266 | 135+ | 157+ | 60°C/1 mM |
| (mut.) | R: TGCAGCCAGCCAATGATGTT | 109+22 | 109 | |||||
| T1019N | SSCP | 231 | 25°C/2h | F: GACCATACTGACCTTGACCC | 353 | na | na | 60°C/1 mM |
| (mut.) | R: AGACATGGCATATCTAGGCC | |||||||
| R1193S | RFLP | 236 | HhaI | F: TGCAGATCGAGATGAACAAG | 295 | 193+80 | 273+22 | 60°C/1 mM |
| (mut.) | R: GTGGGATTAGGAAGTTGGA | +22 | ||||||
| E1426K | RFLP | 236 | TaqI | F: ATCCTCCCCACCCTCTGC | 299 | 140+86 | 213+86 | 60°C/1 mM |
| (mut.) | R: GAGGATGGCTCTGGCCTCT | +73 | ||||||
| R1634C | RFLP | 234 | PvuIIa | F: AGCTCAGCCACGCCAGC | 135 | 135 | 117+18 | 60°C/1 mM |
| (mut.) | R: CCTGTATCAAGACACTACTGCTTACG | |||||||
| VCL gene | ||||||||
| P943A | RFLP | na | BslI | F: TCCTAGGCAGGCTTTGGTTA | 237 | 189+48 | 237 | 60°C/2 mM |
| (polym.) | R: TTGGATGGCATTAACAGCAG | |||||||
SSCP conditions referred to electrophoresis temperature and time (hours).
na: not applicable; mut, mutation; polym, polymorphism; F, forward; R, reverse; bp, base pair.
aArtificial restriction site.
Genotyping conditions for identified mutations and non-synonymous molecular variants
| Molecular variant | Method | Number of controls genotyped | Restriction enzyme; SSCP conditions | PCR primer sequence (5′→3′) | Size (bp) | Allele sizes (bp) | PCR conditions (Temp/[MgCl2]) | |
|---|---|---|---|---|---|---|---|---|
| MYH7 gene | ||||||||
| I201T | SSCP | 224 | 10°C/1h | F: CACTGCCCAATAAGCCCC | 165 | na | na | 65°C/2 mM |
| (mut.) | R: CAGGACCTTGGAGGGCAG | |||||||
| T412N | RFLP | 232 | StyI | F: TTACAGGCATGAACCACACACC | 266 | 169+97 | 266 | 60°C/1 mM |
| (mut.) | R: GTGAACTTGAAAACTCTCATCCC | |||||||
| A550 V | RFLP | 206 | HaeIII | F: GAGCAGAATCCATGTCACCT | 266 | 135+ | 157+ | 60°C/1 mM |
| (mut.) | R: TGCAGCCAGCCAATGATGTT | 109+22 | 109 | |||||
| T1019N | SSCP | 231 | 25°C/2h | F: GACCATACTGACCTTGACCC | 353 | na | na | 60°C/1 mM |
| (mut.) | R: AGACATGGCATATCTAGGCC | |||||||
| R1193S | RFLP | 236 | HhaI | F: TGCAGATCGAGATGAACAAG | 295 | 193+80 | 273+22 | 60°C/1 mM |
| (mut.) | R: GTGGGATTAGGAAGTTGGA | +22 | ||||||
| E1426K | RFLP | 236 | TaqI | F: ATCCTCCCCACCCTCTGC | 299 | 140+86 | 213+86 | 60°C/1 mM |
| (mut.) | R: GAGGATGGCTCTGGCCTCT | +73 | ||||||
| R1634C | RFLP | 234 | PvuIIa | F: AGCTCAGCCACGCCAGC | 135 | 135 | 117+18 | 60°C/1 mM |
| (mut.) | R: CCTGTATCAAGACACTACTGCTTACG | |||||||
| VCL gene | ||||||||
| P943A | RFLP | na | BslI | F: TCCTAGGCAGGCTTTGGTTA | 237 | 189+48 | 237 | 60°C/2 mM |
| (polym.) | R: TTGGATGGCATTAACAGCAG | |||||||
| Molecular variant | Method | Number of controls genotyped | Restriction enzyme; SSCP conditions | PCR primer sequence (5′→3′) | Size (bp) | Allele sizes (bp) | PCR conditions (Temp/[MgCl2]) | |
|---|---|---|---|---|---|---|---|---|
| MYH7 gene | ||||||||
| I201T | SSCP | 224 | 10°C/1h | F: CACTGCCCAATAAGCCCC | 165 | na | na | 65°C/2 mM |
| (mut.) | R: CAGGACCTTGGAGGGCAG | |||||||
| T412N | RFLP | 232 | StyI | F: TTACAGGCATGAACCACACACC | 266 | 169+97 | 266 | 60°C/1 mM |
| (mut.) | R: GTGAACTTGAAAACTCTCATCCC | |||||||
| A550 V | RFLP | 206 | HaeIII | F: GAGCAGAATCCATGTCACCT | 266 | 135+ | 157+ | 60°C/1 mM |
| (mut.) | R: TGCAGCCAGCCAATGATGTT | 109+22 | 109 | |||||
| T1019N | SSCP | 231 | 25°C/2h | F: GACCATACTGACCTTGACCC | 353 | na | na | 60°C/1 mM |
| (mut.) | R: AGACATGGCATATCTAGGCC | |||||||
| R1193S | RFLP | 236 | HhaI | F: TGCAGATCGAGATGAACAAG | 295 | 193+80 | 273+22 | 60°C/1 mM |
| (mut.) | R: GTGGGATTAGGAAGTTGGA | +22 | ||||||
| E1426K | RFLP | 236 | TaqI | F: ATCCTCCCCACCCTCTGC | 299 | 140+86 | 213+86 | 60°C/1 mM |
| (mut.) | R: GAGGATGGCTCTGGCCTCT | +73 | ||||||
| R1634C | RFLP | 234 | PvuIIa | F: AGCTCAGCCACGCCAGC | 135 | 135 | 117+18 | 60°C/1 mM |
| (mut.) | R: CCTGTATCAAGACACTACTGCTTACG | |||||||
| VCL gene | ||||||||
| P943A | RFLP | na | BslI | F: TCCTAGGCAGGCTTTGGTTA | 237 | 189+48 | 237 | 60°C/2 mM |
| (polym.) | R: TTGGATGGCATTAACAGCAG | |||||||
SSCP conditions referred to electrophoresis temperature and time (hours).
na: not applicable; mut, mutation; polym, polymorphism; F, forward; R, reverse; bp, base pair.
aArtificial restriction site.
Identified polymorphisms in coding sequence
| Variant | Nucleotide change | Rare allele frequency (%) |
|---|---|---|
| Polymorphism identified in VCL exon 19 (Acc: NM_014000) | ||
| Gly938Gly | 2908C/G | 0.25 |
| Pro943Ala | 2921C/G | <0.01 |
| Polymorphism identified in TNNT2 (Acc: NM_000364) | ||
| Ser69Ser | 306G/A | 0.04 |
| Ile106Ile | 417T/C | 0.30 |
| Polymorphism identified in PLN (Acc: NM_002667) | ||
| Arg9Arg | 208C/G | 0.04 |
| Polymorphism identified in MYH7 (Acc: AJ238393) | ||
| Thr63Thr | 4557C/T | 0.48 |
| Ala199Ala | 6293A/G | 0.01 |
| Phe244Phe | 6511T/C | 0.26 |
| Asp325Asp | 7510C/T | 0.02 |
| Gly354Gly | 8243C/T | 0.09 |
| Lys365Lys | 8276G/A | 0.16 |
| Asp376Asp | 8309C/T | 0.09 |
| Glu535Glu | 10216A/G | 0.01 |
| Asp778Asp | 12716C/T | 0.01 |
| Asn923Asn | 14029C/T | 0.01 |
| Ile989Ile | 14410T/C | 0.40 |
| Ala1012Ala | 14479C/T | 0.01 |
| Ala1051Ala | 15778G/A | 0.01 |
| Leu1343Leu | 19749T/G | 0.01 |
| Ser1491Cys | 20845C/G | 0.01 |
| Thr1522Thr | 21088T/C | 0.01 |
| Ala1702Ala | 22355G/A | 0.13 |
| Glu1799Glu | 22878A/G | 0.01 |
| Variant | Nucleotide change | Rare allele frequency (%) |
|---|---|---|
| Polymorphism identified in VCL exon 19 (Acc: NM_014000) | ||
| Gly938Gly | 2908C/G | 0.25 |
| Pro943Ala | 2921C/G | <0.01 |
| Polymorphism identified in TNNT2 (Acc: NM_000364) | ||
| Ser69Ser | 306G/A | 0.04 |
| Ile106Ile | 417T/C | 0.30 |
| Polymorphism identified in PLN (Acc: NM_002667) | ||
| Arg9Arg | 208C/G | 0.04 |
| Polymorphism identified in MYH7 (Acc: AJ238393) | ||
| Thr63Thr | 4557C/T | 0.48 |
| Ala199Ala | 6293A/G | 0.01 |
| Phe244Phe | 6511T/C | 0.26 |
| Asp325Asp | 7510C/T | 0.02 |
| Gly354Gly | 8243C/T | 0.09 |
| Lys365Lys | 8276G/A | 0.16 |
| Asp376Asp | 8309C/T | 0.09 |
| Glu535Glu | 10216A/G | 0.01 |
| Asp778Asp | 12716C/T | 0.01 |
| Asn923Asn | 14029C/T | 0.01 |
| Ile989Ile | 14410T/C | 0.40 |
| Ala1012Ala | 14479C/T | 0.01 |
| Ala1051Ala | 15778G/A | 0.01 |
| Leu1343Leu | 19749T/G | 0.01 |
| Ser1491Cys | 20845C/G | 0.01 |
| Thr1522Thr | 21088T/C | 0.01 |
| Ala1702Ala | 22355G/A | 0.13 |
| Glu1799Glu | 22878A/G | 0.01 |
Nucleotide change referred to indicated accession numbers (Acc).
Identified polymorphisms in coding sequence
| Variant | Nucleotide change | Rare allele frequency (%) |
|---|---|---|
| Polymorphism identified in VCL exon 19 (Acc: NM_014000) | ||
| Gly938Gly | 2908C/G | 0.25 |
| Pro943Ala | 2921C/G | <0.01 |
| Polymorphism identified in TNNT2 (Acc: NM_000364) | ||
| Ser69Ser | 306G/A | 0.04 |
| Ile106Ile | 417T/C | 0.30 |
| Polymorphism identified in PLN (Acc: NM_002667) | ||
| Arg9Arg | 208C/G | 0.04 |
| Polymorphism identified in MYH7 (Acc: AJ238393) | ||
| Thr63Thr | 4557C/T | 0.48 |
| Ala199Ala | 6293A/G | 0.01 |
| Phe244Phe | 6511T/C | 0.26 |
| Asp325Asp | 7510C/T | 0.02 |
| Gly354Gly | 8243C/T | 0.09 |
| Lys365Lys | 8276G/A | 0.16 |
| Asp376Asp | 8309C/T | 0.09 |
| Glu535Glu | 10216A/G | 0.01 |
| Asp778Asp | 12716C/T | 0.01 |
| Asn923Asn | 14029C/T | 0.01 |
| Ile989Ile | 14410T/C | 0.40 |
| Ala1012Ala | 14479C/T | 0.01 |
| Ala1051Ala | 15778G/A | 0.01 |
| Leu1343Leu | 19749T/G | 0.01 |
| Ser1491Cys | 20845C/G | 0.01 |
| Thr1522Thr | 21088T/C | 0.01 |
| Ala1702Ala | 22355G/A | 0.13 |
| Glu1799Glu | 22878A/G | 0.01 |
| Variant | Nucleotide change | Rare allele frequency (%) |
|---|---|---|
| Polymorphism identified in VCL exon 19 (Acc: NM_014000) | ||
| Gly938Gly | 2908C/G | 0.25 |
| Pro943Ala | 2921C/G | <0.01 |
| Polymorphism identified in TNNT2 (Acc: NM_000364) | ||
| Ser69Ser | 306G/A | 0.04 |
| Ile106Ile | 417T/C | 0.30 |
| Polymorphism identified in PLN (Acc: NM_002667) | ||
| Arg9Arg | 208C/G | 0.04 |
| Polymorphism identified in MYH7 (Acc: AJ238393) | ||
| Thr63Thr | 4557C/T | 0.48 |
| Ala199Ala | 6293A/G | 0.01 |
| Phe244Phe | 6511T/C | 0.26 |
| Asp325Asp | 7510C/T | 0.02 |
| Gly354Gly | 8243C/T | 0.09 |
| Lys365Lys | 8276G/A | 0.16 |
| Asp376Asp | 8309C/T | 0.09 |
| Glu535Glu | 10216A/G | 0.01 |
| Asp778Asp | 12716C/T | 0.01 |
| Asn923Asn | 14029C/T | 0.01 |
| Ile989Ile | 14410T/C | 0.40 |
| Ala1012Ala | 14479C/T | 0.01 |
| Ala1051Ala | 15778G/A | 0.01 |
| Leu1343Leu | 19749T/G | 0.01 |
| Ser1491Cys | 20845C/G | 0.01 |
| Thr1522Thr | 21088T/C | 0.01 |
| Ala1702Ala | 22355G/A | 0.13 |
| Glu1799Glu | 22878A/G | 0.01 |
Nucleotide change referred to indicated accession numbers (Acc).
Clinical features of DCM mutation carriers
| Subject | Age (years)/sex | Age at diagnosis (years) | NYHA class | ECG | LVEDD (mm) | EF (%) | IVS (mm) | Muscular clin./CPK/biopsy | Clinical status | Comments |
|---|---|---|---|---|---|---|---|---|---|---|
| Beta myosin mutations | ||||||||||
| Family A | ||||||||||
| I-2 | 89/M | 89 | II | SR, cLBB | 65a | 40 | 10 | NL/NA/NA | Affected | Diagnosis during inquest |
| II-1 | 56/W | NA | I | SR | 54a | 60 | 8 | NL/NA/NA | Unknown | |
| II-3 | 53/W | 47 | II | SR | 61a | 26 | 8 | NL/NL/NL | Affected | |
| III-1 | 22/W | 22 | I | SR | 56a | 46 | 6 | NL/NA/NA | Affected | |
| Family B | ||||||||||
| II-1 | 43/M | 33 | IV | SR, iLBB | 93a | 20 | 7 | NL/NA/NA | Affected | HT at 39 years |
| II-2 | 49/M | 47 | III | SR, iLBB | 78a | 16 | NA | NL/NL/NA | Affected | Cardiac death at 52 years |
| II-3 | 51/M | NA | NA | SR, cLBB | 73a | 20 | 10 | NL/NA/NA | Affected | No DNA |
| Family C | ||||||||||
| II-1 | 54/W | 52 | III | SR, iLBB, PVB | 58a | 38 | 10.5 | NL/NL/NL | Affected | |
| II-3 | 58/W | NA | IV | NA | Enlarged | Low | NA | NA/NA/NA | Affected | HT at 49 years, DCM on histopathology, death at 58 years, obligate mutation carrier |
| II-6 | 58/W | 53 | III | SR, Q waves | 61a | 20 | 9 | NL/NL/NA | Unknown | MI and LVA occlusion, HT at 60 years |
| II-8 | 55/M | 47 | IV | SR, cLBB, Q waves | 64a | 20 | 10 | NL/NL/NA | Unknown | MI, CABG, cardiac death at 57 years |
| III-1 | 37/W | — | I | SR | 52 | 59 | 8 | NL/NL/NA | Healthy | |
| III-2 | 42/W | — | I | SR | 54a | 55 | 8 | NL/NL/NA | Healthy | Surgery for IAD at 15 years |
| III-3 | 12/M | — | I | SR | NA | NA | NA | NL/NL/NA | Healthy | |
| III-4 | 25/W | — | I | SR | 46 | 76 | 6.5 | NL/NA/NA | Healthy | |
| III-5 | 23/W | — | I | SR | 45 | 86 | 6.5 | NL/NL/NA | Healthy | |
| Family D | ||||||||||
| II-2 | 56/W | — | I | SR | 53 | 45 | 9 | NL/NA/NA | Unknown | |
| II-4 | 65/W | 58 | II | SR | 59a | 35 | 8 | NL/NA/NA | Affected | |
| II-5 | 60/M | — | I | SR | 56a | 57 | 9 | NL/NA/NA | Unknown | |
| II-6 | 59/M | — | I | SR, iRBB | 55 | 67 | 11 | NL/NA/NA | Healthy | |
| II-7 | 55/M | 49 | II | SR, cLBB | 57a | 45 | 9 | NL/NA/NA | Affected | |
| II-9 | 53/M | — | I | SR | 60a | 55 | 8 | NL/NA/NA | Unknown | |
| II-10 | 45/M | 38 | I | SR, PVB, AVB II | 68a | 30 | 9 | NL/NA/NA | Affected | Deafness |
| III-2 | 32/M | 31 | I | SR | 60a | 43 | 9 | NL/NA/NA | Affected | Obligate mutation carrier |
| III-7 | 9/M | — | I | SR | 42 | 63 | 5.5 | NL/NA/NA | Healthy | |
| IV-1 | 10/M | — | I | SR | 43 | 58 | 7 | NL/NA/NA | Healthy | |
| Family E | ||||||||||
| I-1 | 78/W | 77 | II | SR, iLBB | 62a | 23 | 8.5 | NL/NL/NA | Affected | |
| II-1 | 55/W | NA | NA | SR | 53 | 41 | 8 | NL/NL/NA | Affected | Obligate mutation carrier |
| III-1 | 28/W | 27 | II | SR | 66a | 35 | 6 | NL/NL/NA | Affected | |
| Sporadic A | ||||||||||
| II-1 | 40/M | 39 | II | SR, iLBB | 79a | 25 | 8 | NL/NL/NL | Affected | |
| Sporadic B | ||||||||||
| II-1 | 65/M | 62 | II | SR, PVB, iLBB | 58a | 38 | 11 | NL/NL/NA | Affected | Hypertension |
| T troponin mutations | ||||||||||
| Family F | ||||||||||
| II-2 | 77/F | 54 | II | SR, cLBB, VTns | 62a | 30 | 9 | NL/NL/NA | Affected | |
| III-9 | 42/F | 34 | III | SR, iLBB | 63a | 33 | 8 | NL/NL/NA | Affected | Diagnosis during pregnancy (dyspnoea) |
| IV-3 | 11/M | 2 months | II | SR | 25a at 2 month | 44 | NL | NL/NA/NA | Affected | Improvment of EF: 58% at 4 years |
| Sporadic C | ||||||||||
| II-1 | 1.5 month | 1.5 month | IV | SR, iLBB | Increased | <30 | NL | NL/NA/NA | Affected | CHF/death at 1.5 month |
| Subject | Age (years)/sex | Age at diagnosis (years) | NYHA class | ECG | LVEDD (mm) | EF (%) | IVS (mm) | Muscular clin./CPK/biopsy | Clinical status | Comments |
|---|---|---|---|---|---|---|---|---|---|---|
| Beta myosin mutations | ||||||||||
| Family A | ||||||||||
| I-2 | 89/M | 89 | II | SR, cLBB | 65a | 40 | 10 | NL/NA/NA | Affected | Diagnosis during inquest |
| II-1 | 56/W | NA | I | SR | 54a | 60 | 8 | NL/NA/NA | Unknown | |
| II-3 | 53/W | 47 | II | SR | 61a | 26 | 8 | NL/NL/NL | Affected | |
| III-1 | 22/W | 22 | I | SR | 56a | 46 | 6 | NL/NA/NA | Affected | |
| Family B | ||||||||||
| II-1 | 43/M | 33 | IV | SR, iLBB | 93a | 20 | 7 | NL/NA/NA | Affected | HT at 39 years |
| II-2 | 49/M | 47 | III | SR, iLBB | 78a | 16 | NA | NL/NL/NA | Affected | Cardiac death at 52 years |
| II-3 | 51/M | NA | NA | SR, cLBB | 73a | 20 | 10 | NL/NA/NA | Affected | No DNA |
| Family C | ||||||||||
| II-1 | 54/W | 52 | III | SR, iLBB, PVB | 58a | 38 | 10.5 | NL/NL/NL | Affected | |
| II-3 | 58/W | NA | IV | NA | Enlarged | Low | NA | NA/NA/NA | Affected | HT at 49 years, DCM on histopathology, death at 58 years, obligate mutation carrier |
| II-6 | 58/W | 53 | III | SR, Q waves | 61a | 20 | 9 | NL/NL/NA | Unknown | MI and LVA occlusion, HT at 60 years |
| II-8 | 55/M | 47 | IV | SR, cLBB, Q waves | 64a | 20 | 10 | NL/NL/NA | Unknown | MI, CABG, cardiac death at 57 years |
| III-1 | 37/W | — | I | SR | 52 | 59 | 8 | NL/NL/NA | Healthy | |
| III-2 | 42/W | — | I | SR | 54a | 55 | 8 | NL/NL/NA | Healthy | Surgery for IAD at 15 years |
| III-3 | 12/M | — | I | SR | NA | NA | NA | NL/NL/NA | Healthy | |
| III-4 | 25/W | — | I | SR | 46 | 76 | 6.5 | NL/NA/NA | Healthy | |
| III-5 | 23/W | — | I | SR | 45 | 86 | 6.5 | NL/NL/NA | Healthy | |
| Family D | ||||||||||
| II-2 | 56/W | — | I | SR | 53 | 45 | 9 | NL/NA/NA | Unknown | |
| II-4 | 65/W | 58 | II | SR | 59a | 35 | 8 | NL/NA/NA | Affected | |
| II-5 | 60/M | — | I | SR | 56a | 57 | 9 | NL/NA/NA | Unknown | |
| II-6 | 59/M | — | I | SR, iRBB | 55 | 67 | 11 | NL/NA/NA | Healthy | |
| II-7 | 55/M | 49 | II | SR, cLBB | 57a | 45 | 9 | NL/NA/NA | Affected | |
| II-9 | 53/M | — | I | SR | 60a | 55 | 8 | NL/NA/NA | Unknown | |
| II-10 | 45/M | 38 | I | SR, PVB, AVB II | 68a | 30 | 9 | NL/NA/NA | Affected | Deafness |
| III-2 | 32/M | 31 | I | SR | 60a | 43 | 9 | NL/NA/NA | Affected | Obligate mutation carrier |
| III-7 | 9/M | — | I | SR | 42 | 63 | 5.5 | NL/NA/NA | Healthy | |
| IV-1 | 10/M | — | I | SR | 43 | 58 | 7 | NL/NA/NA | Healthy | |
| Family E | ||||||||||
| I-1 | 78/W | 77 | II | SR, iLBB | 62a | 23 | 8.5 | NL/NL/NA | Affected | |
| II-1 | 55/W | NA | NA | SR | 53 | 41 | 8 | NL/NL/NA | Affected | Obligate mutation carrier |
| III-1 | 28/W | 27 | II | SR | 66a | 35 | 6 | NL/NL/NA | Affected | |
| Sporadic A | ||||||||||
| II-1 | 40/M | 39 | II | SR, iLBB | 79a | 25 | 8 | NL/NL/NL | Affected | |
| Sporadic B | ||||||||||
| II-1 | 65/M | 62 | II | SR, PVB, iLBB | 58a | 38 | 11 | NL/NL/NA | Affected | Hypertension |
| T troponin mutations | ||||||||||
| Family F | ||||||||||
| II-2 | 77/F | 54 | II | SR, cLBB, VTns | 62a | 30 | 9 | NL/NL/NA | Affected | |
| III-9 | 42/F | 34 | III | SR, iLBB | 63a | 33 | 8 | NL/NL/NA | Affected | Diagnosis during pregnancy (dyspnoea) |
| IV-3 | 11/M | 2 months | II | SR | 25a at 2 month | 44 | NL | NL/NA/NA | Affected | Improvment of EF: 58% at 4 years |
| Sporadic C | ||||||||||
| II-1 | 1.5 month | 1.5 month | IV | SR, iLBB | Increased | <30 | NL | NL/NA/NA | Affected | CHF/death at 1.5 month |
Age is age at genetic inquest; NYHA, New York Heart Association functional class; LVEDD, left ventricular end diastolic diameter (mm); EF, ejection fraction (%); IVS, interventricular septum thickness (echography); muscular clin., abnormality in clinical muscular testing; CPK, elevated serum creatine kinase level; M, man; W, woman; SR, sinus rhythm; PVB, pre-mature ventricular beats; iLBB or cLBB, incomplete or complete left bundle branch block; rBB, right bundle branch block; AVB II, atrioventricular block grade II; NL, normal; NA, not available; HT, heart transplantation; MI, myocardial infarction; LVA, left ventricular anterior coronary; CABG, coronary artery bypass graft; IAD, interatrial defect; no DNA, no DNA available for genotyping.
aLVEDD above theoretical value according to age and body surface area.
Clinical features of DCM mutation carriers
| Subject | Age (years)/sex | Age at diagnosis (years) | NYHA class | ECG | LVEDD (mm) | EF (%) | IVS (mm) | Muscular clin./CPK/biopsy | Clinical status | Comments |
|---|---|---|---|---|---|---|---|---|---|---|
| Beta myosin mutations | ||||||||||
| Family A | ||||||||||
| I-2 | 89/M | 89 | II | SR, cLBB | 65a | 40 | 10 | NL/NA/NA | Affected | Diagnosis during inquest |
| II-1 | 56/W | NA | I | SR | 54a | 60 | 8 | NL/NA/NA | Unknown | |
| II-3 | 53/W | 47 | II | SR | 61a | 26 | 8 | NL/NL/NL | Affected | |
| III-1 | 22/W | 22 | I | SR | 56a | 46 | 6 | NL/NA/NA | Affected | |
| Family B | ||||||||||
| II-1 | 43/M | 33 | IV | SR, iLBB | 93a | 20 | 7 | NL/NA/NA | Affected | HT at 39 years |
| II-2 | 49/M | 47 | III | SR, iLBB | 78a | 16 | NA | NL/NL/NA | Affected | Cardiac death at 52 years |
| II-3 | 51/M | NA | NA | SR, cLBB | 73a | 20 | 10 | NL/NA/NA | Affected | No DNA |
| Family C | ||||||||||
| II-1 | 54/W | 52 | III | SR, iLBB, PVB | 58a | 38 | 10.5 | NL/NL/NL | Affected | |
| II-3 | 58/W | NA | IV | NA | Enlarged | Low | NA | NA/NA/NA | Affected | HT at 49 years, DCM on histopathology, death at 58 years, obligate mutation carrier |
| II-6 | 58/W | 53 | III | SR, Q waves | 61a | 20 | 9 | NL/NL/NA | Unknown | MI and LVA occlusion, HT at 60 years |
| II-8 | 55/M | 47 | IV | SR, cLBB, Q waves | 64a | 20 | 10 | NL/NL/NA | Unknown | MI, CABG, cardiac death at 57 years |
| III-1 | 37/W | — | I | SR | 52 | 59 | 8 | NL/NL/NA | Healthy | |
| III-2 | 42/W | — | I | SR | 54a | 55 | 8 | NL/NL/NA | Healthy | Surgery for IAD at 15 years |
| III-3 | 12/M | — | I | SR | NA | NA | NA | NL/NL/NA | Healthy | |
| III-4 | 25/W | — | I | SR | 46 | 76 | 6.5 | NL/NA/NA | Healthy | |
| III-5 | 23/W | — | I | SR | 45 | 86 | 6.5 | NL/NL/NA | Healthy | |
| Family D | ||||||||||
| II-2 | 56/W | — | I | SR | 53 | 45 | 9 | NL/NA/NA | Unknown | |
| II-4 | 65/W | 58 | II | SR | 59a | 35 | 8 | NL/NA/NA | Affected | |
| II-5 | 60/M | — | I | SR | 56a | 57 | 9 | NL/NA/NA | Unknown | |
| II-6 | 59/M | — | I | SR, iRBB | 55 | 67 | 11 | NL/NA/NA | Healthy | |
| II-7 | 55/M | 49 | II | SR, cLBB | 57a | 45 | 9 | NL/NA/NA | Affected | |
| II-9 | 53/M | — | I | SR | 60a | 55 | 8 | NL/NA/NA | Unknown | |
| II-10 | 45/M | 38 | I | SR, PVB, AVB II | 68a | 30 | 9 | NL/NA/NA | Affected | Deafness |
| III-2 | 32/M | 31 | I | SR | 60a | 43 | 9 | NL/NA/NA | Affected | Obligate mutation carrier |
| III-7 | 9/M | — | I | SR | 42 | 63 | 5.5 | NL/NA/NA | Healthy | |
| IV-1 | 10/M | — | I | SR | 43 | 58 | 7 | NL/NA/NA | Healthy | |
| Family E | ||||||||||
| I-1 | 78/W | 77 | II | SR, iLBB | 62a | 23 | 8.5 | NL/NL/NA | Affected | |
| II-1 | 55/W | NA | NA | SR | 53 | 41 | 8 | NL/NL/NA | Affected | Obligate mutation carrier |
| III-1 | 28/W | 27 | II | SR | 66a | 35 | 6 | NL/NL/NA | Affected | |
| Sporadic A | ||||||||||
| II-1 | 40/M | 39 | II | SR, iLBB | 79a | 25 | 8 | NL/NL/NL | Affected | |
| Sporadic B | ||||||||||
| II-1 | 65/M | 62 | II | SR, PVB, iLBB | 58a | 38 | 11 | NL/NL/NA | Affected | Hypertension |
| T troponin mutations | ||||||||||
| Family F | ||||||||||
| II-2 | 77/F | 54 | II | SR, cLBB, VTns | 62a | 30 | 9 | NL/NL/NA | Affected | |
| III-9 | 42/F | 34 | III | SR, iLBB | 63a | 33 | 8 | NL/NL/NA | Affected | Diagnosis during pregnancy (dyspnoea) |
| IV-3 | 11/M | 2 months | II | SR | 25a at 2 month | 44 | NL | NL/NA/NA | Affected | Improvment of EF: 58% at 4 years |
| Sporadic C | ||||||||||
| II-1 | 1.5 month | 1.5 month | IV | SR, iLBB | Increased | <30 | NL | NL/NA/NA | Affected | CHF/death at 1.5 month |
| Subject | Age (years)/sex | Age at diagnosis (years) | NYHA class | ECG | LVEDD (mm) | EF (%) | IVS (mm) | Muscular clin./CPK/biopsy | Clinical status | Comments |
|---|---|---|---|---|---|---|---|---|---|---|
| Beta myosin mutations | ||||||||||
| Family A | ||||||||||
| I-2 | 89/M | 89 | II | SR, cLBB | 65a | 40 | 10 | NL/NA/NA | Affected | Diagnosis during inquest |
| II-1 | 56/W | NA | I | SR | 54a | 60 | 8 | NL/NA/NA | Unknown | |
| II-3 | 53/W | 47 | II | SR | 61a | 26 | 8 | NL/NL/NL | Affected | |
| III-1 | 22/W | 22 | I | SR | 56a | 46 | 6 | NL/NA/NA | Affected | |
| Family B | ||||||||||
| II-1 | 43/M | 33 | IV | SR, iLBB | 93a | 20 | 7 | NL/NA/NA | Affected | HT at 39 years |
| II-2 | 49/M | 47 | III | SR, iLBB | 78a | 16 | NA | NL/NL/NA | Affected | Cardiac death at 52 years |
| II-3 | 51/M | NA | NA | SR, cLBB | 73a | 20 | 10 | NL/NA/NA | Affected | No DNA |
| Family C | ||||||||||
| II-1 | 54/W | 52 | III | SR, iLBB, PVB | 58a | 38 | 10.5 | NL/NL/NL | Affected | |
| II-3 | 58/W | NA | IV | NA | Enlarged | Low | NA | NA/NA/NA | Affected | HT at 49 years, DCM on histopathology, death at 58 years, obligate mutation carrier |
| II-6 | 58/W | 53 | III | SR, Q waves | 61a | 20 | 9 | NL/NL/NA | Unknown | MI and LVA occlusion, HT at 60 years |
| II-8 | 55/M | 47 | IV | SR, cLBB, Q waves | 64a | 20 | 10 | NL/NL/NA | Unknown | MI, CABG, cardiac death at 57 years |
| III-1 | 37/W | — | I | SR | 52 | 59 | 8 | NL/NL/NA | Healthy | |
| III-2 | 42/W | — | I | SR | 54a | 55 | 8 | NL/NL/NA | Healthy | Surgery for IAD at 15 years |
| III-3 | 12/M | — | I | SR | NA | NA | NA | NL/NL/NA | Healthy | |
| III-4 | 25/W | — | I | SR | 46 | 76 | 6.5 | NL/NA/NA | Healthy | |
| III-5 | 23/W | — | I | SR | 45 | 86 | 6.5 | NL/NL/NA | Healthy | |
| Family D | ||||||||||
| II-2 | 56/W | — | I | SR | 53 | 45 | 9 | NL/NA/NA | Unknown | |
| II-4 | 65/W | 58 | II | SR | 59a | 35 | 8 | NL/NA/NA | Affected | |
| II-5 | 60/M | — | I | SR | 56a | 57 | 9 | NL/NA/NA | Unknown | |
| II-6 | 59/M | — | I | SR, iRBB | 55 | 67 | 11 | NL/NA/NA | Healthy | |
| II-7 | 55/M | 49 | II | SR, cLBB | 57a | 45 | 9 | NL/NA/NA | Affected | |
| II-9 | 53/M | — | I | SR | 60a | 55 | 8 | NL/NA/NA | Unknown | |
| II-10 | 45/M | 38 | I | SR, PVB, AVB II | 68a | 30 | 9 | NL/NA/NA | Affected | Deafness |
| III-2 | 32/M | 31 | I | SR | 60a | 43 | 9 | NL/NA/NA | Affected | Obligate mutation carrier |
| III-7 | 9/M | — | I | SR | 42 | 63 | 5.5 | NL/NA/NA | Healthy | |
| IV-1 | 10/M | — | I | SR | 43 | 58 | 7 | NL/NA/NA | Healthy | |
| Family E | ||||||||||
| I-1 | 78/W | 77 | II | SR, iLBB | 62a | 23 | 8.5 | NL/NL/NA | Affected | |
| II-1 | 55/W | NA | NA | SR | 53 | 41 | 8 | NL/NL/NA | Affected | Obligate mutation carrier |
| III-1 | 28/W | 27 | II | SR | 66a | 35 | 6 | NL/NL/NA | Affected | |
| Sporadic A | ||||||||||
| II-1 | 40/M | 39 | II | SR, iLBB | 79a | 25 | 8 | NL/NL/NL | Affected | |
| Sporadic B | ||||||||||
| II-1 | 65/M | 62 | II | SR, PVB, iLBB | 58a | 38 | 11 | NL/NL/NA | Affected | Hypertension |
| T troponin mutations | ||||||||||
| Family F | ||||||||||
| II-2 | 77/F | 54 | II | SR, cLBB, VTns | 62a | 30 | 9 | NL/NL/NA | Affected | |
| III-9 | 42/F | 34 | III | SR, iLBB | 63a | 33 | 8 | NL/NL/NA | Affected | Diagnosis during pregnancy (dyspnoea) |
| IV-3 | 11/M | 2 months | II | SR | 25a at 2 month | 44 | NL | NL/NA/NA | Affected | Improvment of EF: 58% at 4 years |
| Sporadic C | ||||||||||
| II-1 | 1.5 month | 1.5 month | IV | SR, iLBB | Increased | <30 | NL | NL/NA/NA | Affected | CHF/death at 1.5 month |
Age is age at genetic inquest; NYHA, New York Heart Association functional class; LVEDD, left ventricular end diastolic diameter (mm); EF, ejection fraction (%); IVS, interventricular septum thickness (echography); muscular clin., abnormality in clinical muscular testing; CPK, elevated serum creatine kinase level; M, man; W, woman; SR, sinus rhythm; PVB, pre-mature ventricular beats; iLBB or cLBB, incomplete or complete left bundle branch block; rBB, right bundle branch block; AVB II, atrioventricular block grade II; NL, normal; NA, not available; HT, heart transplantation; MI, myocardial infarction; LVA, left ventricular anterior coronary; CABG, coronary artery bypass graft; IAD, interatrial defect; no DNA, no DNA available for genotyping.
aLVEDD above theoretical value according to age and body surface area.
References
Michels VV, Moll PP, Miller FA et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy.
Keeling PJ, Gang Y, Smith G et al. Familial dilated cardiomyopathy in the United Kingdom.
Grünig E, Tasman JA, Kücherer H et al. Frequency and phenotypes of familial dilated cardiomyopathy.
Schonberger J, Seidman CE. Many roads lead to a broken heart: the genetics of dilated cardiomyopathy.
Gerull B, Gramlich M, Atherton J et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy.
Olson TM, Illenberger S, Kishimoto NY et al. Metavinculin mutations alter actin interaction in dilated cardiomyopathy.
Knoll R, Hoshijima M, Hoffman HM et al. The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy.
Schmitt JP, Kamisago M, Asahi M et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban.
Arimura T, Hayashi T, Terada H et al. A Cypher/ZASP mutation associated with dilated cardiomyopathy alters the binding affinity to protein kinase C.
Kamisago M, Sharma SD, DePalma SR et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy.
Daehmlow S, Erdmann J, Knueppel T et al. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy.
Arbustini E, Morbini P, Pilotto A et al. Familial dilated cardiomyopathy: from clinical presentation to molecular genetics.
Tesson F, Sylvius N, Pilotto A et al. Epidemiology of desmin and cardiac actin gene mutations in a European population of dilated cardiomyopathy.
Sylvius N, Duboscq-Bidot L, Bouchier C et al. Mutational analysis of the beta- and delta-sarcoglycan genes in a large number of patients with familial and sporadic dilated cardiomyopathy.
Sebillon P, Bouchier C, Bidot LD et al. Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations.
Mestroni L, Maisch B, McKenna WJ et al. Guidelines for the study of familial dilated cardiomyopathies.
Blair E, Redwood C, de Jesus Oliveira M et al. Mutations of the light meromyosin domain of the beta-myosin heavy chain rod in hypertrophic cardiomyopathy.
Li D, Czernuszewicz GZ, Gonzalez O et al. Novel cardiac troponin T mutation as a cause of familial dilated cardiomyopathy.
Hanson EL, Jakobs PM, Keegan H et al. Cardiac troponin T lysine 210 deletion in a family with dilated cardiomyopathy.
Freeman K, Nakao K, Leinwand LA. Low sequence variation in the gene encoding the human beta-myosin heavy chain.
Arbustini E, Pilotto A, Repetto A et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect-related disease.
Taylor MR, Fain PR, Sinagra G et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations.


{kind=link}
{kind=link}
{kind=link}
{kind=link}