





Abstract
We investigated the presence of a clinical diagnosis of hypertrophic cardiomyopathy (HCM) and of risk factors for sudden cardiac death (SCD) at the first cardiological evaluation after predictive genetic testing in asymptomatic carriers of an MYBPC3 gene mutation.
Two hundred and thirty-five mutation carriers were cardiologically evaluated on the presence of HCM and risk factors. A clinical diagnosis of HCM was made in 53 carriers (22.6%). Disease penetrance at 65 years was incomplete for all types of MYBPC3 gene mutations. Women were affected less often than men (15 and 32% respectively, P = 0.003) and disease penetrance was lower in females than in males (13 and 30% at 50 years, respectively, P = 0.024). One risk factor was present in 87 carriers and 9 had two or more risk factors. Twenty-five carriers (11%) with one or more risk factors and manifest HCM could be at risk for SCD.
At first cardiological evaluation almost one-quarter of asymptomatic carriers was diagnosed with HCM. Risk factors for SCD were frequently present and 11% of carriers could be at risk for SCD. Predictive genetic testing in HCM families and frequent cardiological evaluation on the presence of HCM and risk factors for SCD are justified until advanced age.
Introduction
Hypertrophic cardiomyopathy (HCM) is a common genetic disease affecting at least 1 in 500 persons in the general population.1,2 The clinical diagnosis rests on the presence of a hypertrophied, non-dilated left ventricle on echocardiography (left ventricle wall thickness ≥15 mm or ≥13 mm in relatives of a patient) in the absence of other cardiac or systemic diseases that may cause cardiac hypertrophy, such as aortic valve stenosis and hypertension.3,4
The risk of sudden cardiac death (SCD) is ∼1% annually in unselected HCM patients but increases to 5% or more if risk factors are present.5,6 Several risk factors associated with an elevated risk of SCD in HCM patients have been identified.7 It has been proposed that in patients with one or more risk factor(s) the implantation of an internal cardioverter defibrillator (ICD), a therapy with proven efficacy in the prevention of SCD, should be considered.7–10
Hypertrophic cardiomyopathy is an autosomal dominant trait. Nowadays, a disease causing mutation is detected in about 50–60% of probands in diagnostic molecular testing.11 These mutations are most often found in the genes encoding sarcomeric proteins. DNA-diagnostics in the Netherlands is possible since 1996 in HCM patients and predictive DNA-diagnostics in relatives started in 2001. In the Netherlands the majority of mutations are located in the cardiac myosin-binding protein C (MYBPC3, MIM *600958) gene. This high proportion is mainly caused by the presence of three Dutch founder mutations (35–40%: c.2373_2374insG,12 c.2827C>T and c.2864_2865delCT) in the MYBPC3 gene.
Identification of a disease causing mutation implies the opportunity of systematic predictive genetic testing in relatives (cascade screening). While proven mutation carriers should be referred for cardiological evaluation according to the ACC/ESC guidelines, prognosis-dependent care is difficult.7 Indeed, in carriers of a familial mutation not much is known about: (i) the risk of developing HCM, (ii) the presence of risk factors for SCD in this group, (iii) the association of these risk factors with an increased risk of SCD in carriers, and (iv) whether the type of mutation involved, affects prognosis. Optimal management of mutation carriers is therefore unclear.
In this paper, we report the results of the first cardiological evaluation in a group of asymptomatic relatives with a proven familial mutation in the MYBPC3 gene identified through active cascade screening and we address the clinical diagnosis of HCM, the presence of risk factors for SCD, and the different types of mutations in the MYBPC3 gene.
Methods
Patients
In 83 families with a (probably) pathogenic MYBPC3 gene mutation, we included asymptomatic relatives not known to have HCM with a proven familial MYBPC3 gene mutation from four Universitary Hospitals. Probands and relatives who had been clinically diagnosed with HCM before genetic testing were excluded. The majority of asymptomatic relatives tested for the familial mutation were first-degree relatives of the proband or of an already detected mutation carrier, who had an a priori risk of carriership of 50%. If the connecting first-degree relative had died, second-degree relatives were also tested. All included mutation carriers provided written informed consent for anonymous use of their data for research purposes.
We started this prospective cohort study in 2001 when the first asymptomatic relatives were identified as mutation carrier through cascade screening. Because of the still increasing number of HCM patients in whom DNA-diagnostics is performed and the successive nature of cascade screening of the relatives, the inclusion of asymptomatic relatives shows an increase in time.
Data
From all mutation carriers a family tree was recorded with data on SCD in relatives until the third degree. Carriers were advised to have cardiological evaluation with an ECG, echocardiogram, 24 h ambulatory Holter recording, and an exercise test regularly.7 Clinical parameters from their first cardiological evaluation (often performed in local hospitals in the proximity of their home) after predictive genetic testing were recorded. A clinical diagnosis of HCM was made when on echocardiography the maximal left-ventricular wall thickness was ≥13 mm and/or severe systolic anterior movement of the mitral valve (SAM) was present.4 In children the clinical diagnosis was made when on echocardiography a maximal wall thickness ≥2 SD for their weight was present. All cardiological evaluations were reviewed by the authors.
The following risk factors for SCD were assessed: The cumulative number of risk factors is the number of the above-mentioned six risk factors for SCD that is positive.
Family history of premature SCD. Unexpected non-traumatic premature death within 1 h after the onset of symptoms and without previous severe symptoms in relatives, including unwittnessed unexpected nocturnal death and equivalents like successful resuscitation or appropriate ICD discharge. With respects to the age and degree of kinship and number of the relative(s) involved, we use the definition most used in literature: two relatives with SCD <40 years.13–15
Unexplained syncope. Unexplained syncope, not judged to be neurocardiogenic.
Prior aborted cardiac arrest or sustained ventricular tachycardia (VT). Cardiac arrest (ventricular fibrillation) in history or spontaneously occurring sustained VT at exercise test or Holter recording.
Non-sustained VT (NSVT). One or more runs of three or more consecutive ventricular extrasystoles at a rate of more than 120 b.p.m. lasting for <30 s at exercise test or 24 h ambulatory Holter recording.
Extreme left-ventricular hypertrophy. Maximum left-ventricular wall thickness of 30 mm or more on echocardiography.
Abnormal blood pressure response (ABPR) during upright exercise. A failure of the systolic blood pressure to rise by more than 20 mmHg from baseline values, or a fall of more than 10 mmHg from the maximum blood pressure during upright exercise (treadmill Bruce protocol or bicycle protocol).
Follow-up after the first cardiological visit was on average 3.2 (SD 1.3; median 3.0; interquartile range 2.3) years. Endpoints in follow-up were death, cardiovascular death, SCD, heart transplantation, and appropriate ICD discharge.
Genetic analysis
Mutation analysis in the probands was performed according to a previously published protocol.16 Truncating mutations were defined as (probably) pathogenic based on descriptions in literature, cosegregation with the phenotype, absence in at least 200 controls, the predicted probability of nonsense-mediated mRNA decay and the results of functional assays. Missense mutations were defined as (probably) pathogenic based on descriptions in literature, cosegregation with the phenotype, absence in at least 200 controls, evolutionary conservation of the amino acid, and chemical differences in the amino acid.
In this study, we distinguished three types of MYBPC3 gene mutations: the c.2373_2374insG truncating mutation, which is the most prevalent Dutch founder mutation, other truncating mutations (including splice site mutations) and missense mutations. The different MYBPC3 gene mutations in the asymptomatic mutation carriers are described in Table 1.
The different MYBPC3 gene mutations in asymptomatic mutation carriers
| Mutation | Number of asymptomatic relatives with this mutation | |
|---|---|---|
| Base pair notation | Amino acid notation | |
| Truncating mutations | ||
| c.927-2A>G | —a | 8 |
| c.932C>A | p.Ser311X | 1 |
| c.2373_2374insGb | p.Trp729fsX17 | 162 |
| c.2827C>T | p.Arg943X | 18 |
| c.2864_2865delCT | p.Pro955fsX95 | 2 |
| c.2893C>T | p.Gln965X | 5 |
| c.2905C>T | p.Gln696X | 3 |
| c.3258_3259insGG | p.Lys1087fsX101 | 10 |
| c.3776delA | p.Gln1259fsX | 3 |
| Missense mutations | ||
| c.481C>T | p.Pro161Ser | 4 |
| c.1483C>G | p.Arg495Gly | 2 |
| c.1484G>A | p.Arg495Gln | 10 |
| c.3332A>G | p.Glu1111Gly | 3 |
| c.3640T>C | p.Trp1214Arg | 4 |
| Mutation | Number of asymptomatic relatives with this mutation | |
|---|---|---|
| Base pair notation | Amino acid notation | |
| Truncating mutations | ||
| c.927-2A>G | —a | 8 |
| c.932C>A | p.Ser311X | 1 |
| c.2373_2374insGb | p.Trp729fsX17 | 162 |
| c.2827C>T | p.Arg943X | 18 |
| c.2864_2865delCT | p.Pro955fsX95 | 2 |
| c.2893C>T | p.Gln965X | 5 |
| c.2905C>T | p.Gln696X | 3 |
| c.3258_3259insGG | p.Lys1087fsX101 | 10 |
| c.3776delA | p.Gln1259fsX | 3 |
| Missense mutations | ||
| c.481C>T | p.Pro161Ser | 4 |
| c.1483C>G | p.Arg495Gly | 2 |
| c.1484G>A | p.Arg495Gln | 10 |
| c.3332A>G | p.Glu1111Gly | 3 |
| c.3640T>C | p.Trp1214Arg | 4 |
aNot applicable for this splice site mutation.
bDutch founder mutation.
The different MYBPC3 gene mutations in asymptomatic mutation carriers
| Mutation | Number of asymptomatic relatives with this mutation | |
|---|---|---|
| Base pair notation | Amino acid notation | |
| Truncating mutations | ||
| c.927-2A>G | —a | 8 |
| c.932C>A | p.Ser311X | 1 |
| c.2373_2374insGb | p.Trp729fsX17 | 162 |
| c.2827C>T | p.Arg943X | 18 |
| c.2864_2865delCT | p.Pro955fsX95 | 2 |
| c.2893C>T | p.Gln965X | 5 |
| c.2905C>T | p.Gln696X | 3 |
| c.3258_3259insGG | p.Lys1087fsX101 | 10 |
| c.3776delA | p.Gln1259fsX | 3 |
| Missense mutations | ||
| c.481C>T | p.Pro161Ser | 4 |
| c.1483C>G | p.Arg495Gly | 2 |
| c.1484G>A | p.Arg495Gln | 10 |
| c.3332A>G | p.Glu1111Gly | 3 |
| c.3640T>C | p.Trp1214Arg | 4 |
| Mutation | Number of asymptomatic relatives with this mutation | |
|---|---|---|
| Base pair notation | Amino acid notation | |
| Truncating mutations | ||
| c.927-2A>G | —a | 8 |
| c.932C>A | p.Ser311X | 1 |
| c.2373_2374insGb | p.Trp729fsX17 | 162 |
| c.2827C>T | p.Arg943X | 18 |
| c.2864_2865delCT | p.Pro955fsX95 | 2 |
| c.2893C>T | p.Gln965X | 5 |
| c.2905C>T | p.Gln696X | 3 |
| c.3258_3259insGG | p.Lys1087fsX101 | 10 |
| c.3776delA | p.Gln1259fsX | 3 |
| Missense mutations | ||
| c.481C>T | p.Pro161Ser | 4 |
| c.1483C>G | p.Arg495Gly | 2 |
| c.1484G>A | p.Arg495Gln | 10 |
| c.3332A>G | p.Glu1111Gly | 3 |
| c.3640T>C | p.Trp1214Arg | 4 |
aNot applicable for this splice site mutation.
bDutch founder mutation.
Statistical analysis
Data were analysed with SPSS (version 15.0) statistical software. Data are expressed as means (SD) or as a frequency. Student's t-test or one-way ANOVA was used for the comparison of normally distributed continuous variables, non-parametric methods for not-normally distributed continuous variables, and Pearson's χ2 for comparisons between dichotomous variables. Kaplan–Meier analysis was used to describe the relationship between the clinical diagnosis of HCM (event) at the first cardiological evaluation and the carrier's age at that time (time to event). Carriers without a clinical diagnosis of HCM were censored after time (age) of first evaluation. Disease manifestations by type of MYBPC3 gene mutation and by gender were compared using the log-rank test. A two sided P-value <0.05 was considered to indicate statistical significance.
Results
Clinical parameters
The mean age of 235 mutation carriers was 39.7 (SD 17.9, range 2-86) years at first cardiological visit; 108 (46%) were men. Clinical parameters are summarized in Table 2.
Clinical parameters of 235 mutation carriers at first cardiological evaluation
| Clinical parameters | All mutation carriers | Mutation carriers with clinical HCM | Mutation carriers without clinical HCM |
|---|---|---|---|
| Age (years) | 39.7 ± 17.9 (235) | 47.0 ± 19.0 (53) | 37.6 ± 17.1 (182)* |
| Male | 108 (235, 46.0%) | 34 (53, 64.2%) | 74 (182, 40.7%)* |
| Clinical diagnosis of HCM | 53 (235, 22.6%) | ||
| Palpitations | 49 (233, 21.0%) | 7 (51, 13.7%) | 42 (182, 23.1%) |
| Chest pain | 14 (235, 6.0%) | 5 (53, 9.4%) | 9 (182, 4.9%) |
| Atrial fibrillation | 1 (235, 0.4%) | 0 (53, 0.0%) | 1 (182, 0.5%) |
| MYBPC3 gene mutation type | |||
| c.2373_2374insG | 162 (235, 68.9%) | 42 (53, 79.2%) | 120 (182, 65.9%) |
| Other truncating mutations | 50 (235, 21.3%) | 8 (53, 15.1%) | 42 (182, 23.1%) |
| Missense mutations | 23 (235, 9.8%) | 3 (53, 5.7%) | 20 (182, 11.0%) |
| Risk factors for SCD | |||
| Extreme left-ventricular hypertrophy | 4 (235, 1.7%) | 4 (53, 7.5%) | 0 (182, 0.0%)** |
| Non-sustained VT | 16 (159, 10.1%) | 9 (35, 25.7%) | 7 (124, 5.6%)** |
| <50 years | 7 (119, 5.9%) | 4 (21, 19.0%) | 3 (98, 3.1%)* |
| ≥50 years | 9 (40, 22.5%) | 5 (14, 35.7%) | 4 (26, 15.4%) |
| Abnormal blood pressure response | 26 (160, 16.3%) | 7 (38, 18.4%) | 19 (122, 15.6%) |
| <50 years | 20 (117, 17.1%) | 15 (94, 16.0%) | 5 (23, 21.7%) |
| ≥50 years | 6 (43, 14.0%) | 4 (28, 14.3%) | 2 (15, 13.3%) |
| Previous cardiac arrest or VT | 1 (235, 0.4%) | 1 (53, 1.9%) | 0 (182, 0.0%) |
| Unexplained syncope | 12 (235, 5.1%) | 3 (53, 5.7%) | 9 (182, 4.9%) |
| Family history of SCD | 46 (235, 19.6%) | 6 (53, 11.3%) | 40 (182, 22.0%) |
| Number of risk factors for SCD | 0.45 ± 0.57 (235) | 0.57 ± 0.67 (53) | 0.41 ± 0.54 (182) |
| 0 risk factors | 139 (235, 59.1%) | 28 (53, 52.8%) | 111 (182, 61.0%) |
| 1 risk factors | 87 (235, 37.0%) | 20 (53, 37.7%) | 67 (182, 36.8%) |
| ≥2 risk factors | 9 (235, 3.8%) | 5 (53, 9.4%) | 4 (182, 2.2%) |
| Clinical parameters | All mutation carriers | Mutation carriers with clinical HCM | Mutation carriers without clinical HCM |
|---|---|---|---|
| Age (years) | 39.7 ± 17.9 (235) | 47.0 ± 19.0 (53) | 37.6 ± 17.1 (182)* |
| Male | 108 (235, 46.0%) | 34 (53, 64.2%) | 74 (182, 40.7%)* |
| Clinical diagnosis of HCM | 53 (235, 22.6%) | ||
| Palpitations | 49 (233, 21.0%) | 7 (51, 13.7%) | 42 (182, 23.1%) |
| Chest pain | 14 (235, 6.0%) | 5 (53, 9.4%) | 9 (182, 4.9%) |
| Atrial fibrillation | 1 (235, 0.4%) | 0 (53, 0.0%) | 1 (182, 0.5%) |
| MYBPC3 gene mutation type | |||
| c.2373_2374insG | 162 (235, 68.9%) | 42 (53, 79.2%) | 120 (182, 65.9%) |
| Other truncating mutations | 50 (235, 21.3%) | 8 (53, 15.1%) | 42 (182, 23.1%) |
| Missense mutations | 23 (235, 9.8%) | 3 (53, 5.7%) | 20 (182, 11.0%) |
| Risk factors for SCD | |||
| Extreme left-ventricular hypertrophy | 4 (235, 1.7%) | 4 (53, 7.5%) | 0 (182, 0.0%)** |
| Non-sustained VT | 16 (159, 10.1%) | 9 (35, 25.7%) | 7 (124, 5.6%)** |
| <50 years | 7 (119, 5.9%) | 4 (21, 19.0%) | 3 (98, 3.1%)* |
| ≥50 years | 9 (40, 22.5%) | 5 (14, 35.7%) | 4 (26, 15.4%) |
| Abnormal blood pressure response | 26 (160, 16.3%) | 7 (38, 18.4%) | 19 (122, 15.6%) |
| <50 years | 20 (117, 17.1%) | 15 (94, 16.0%) | 5 (23, 21.7%) |
| ≥50 years | 6 (43, 14.0%) | 4 (28, 14.3%) | 2 (15, 13.3%) |
| Previous cardiac arrest or VT | 1 (235, 0.4%) | 1 (53, 1.9%) | 0 (182, 0.0%) |
| Unexplained syncope | 12 (235, 5.1%) | 3 (53, 5.7%) | 9 (182, 4.9%) |
| Family history of SCD | 46 (235, 19.6%) | 6 (53, 11.3%) | 40 (182, 22.0%) |
| Number of risk factors for SCD | 0.45 ± 0.57 (235) | 0.57 ± 0.67 (53) | 0.41 ± 0.54 (182) |
| 0 risk factors | 139 (235, 59.1%) | 28 (53, 52.8%) | 111 (182, 61.0%) |
| 1 risk factors | 87 (235, 37.0%) | 20 (53, 37.7%) | 67 (182, 36.8%) |
| ≥2 risk factors | 9 (235, 3.8%) | 5 (53, 9.4%) | 4 (182, 2.2%) |
Data are mean ± SD or number and proportion (%).
HCM, hypertrophic cardiomyopathy; SCD, sudden cardiac death; VT, ventricular tachycardia.
Significant differences between carriers with and without a clinical diagnosis of HCM: *P-value < 0.01, **P-value < 0.001.
Clinical parameters of 235 mutation carriers at first cardiological evaluation
| Clinical parameters | All mutation carriers | Mutation carriers with clinical HCM | Mutation carriers without clinical HCM |
|---|---|---|---|
| Age (years) | 39.7 ± 17.9 (235) | 47.0 ± 19.0 (53) | 37.6 ± 17.1 (182)* |
| Male | 108 (235, 46.0%) | 34 (53, 64.2%) | 74 (182, 40.7%)* |
| Clinical diagnosis of HCM | 53 (235, 22.6%) | ||
| Palpitations | 49 (233, 21.0%) | 7 (51, 13.7%) | 42 (182, 23.1%) |
| Chest pain | 14 (235, 6.0%) | 5 (53, 9.4%) | 9 (182, 4.9%) |
| Atrial fibrillation | 1 (235, 0.4%) | 0 (53, 0.0%) | 1 (182, 0.5%) |
| MYBPC3 gene mutation type | |||
| c.2373_2374insG | 162 (235, 68.9%) | 42 (53, 79.2%) | 120 (182, 65.9%) |
| Other truncating mutations | 50 (235, 21.3%) | 8 (53, 15.1%) | 42 (182, 23.1%) |
| Missense mutations | 23 (235, 9.8%) | 3 (53, 5.7%) | 20 (182, 11.0%) |
| Risk factors for SCD | |||
| Extreme left-ventricular hypertrophy | 4 (235, 1.7%) | 4 (53, 7.5%) | 0 (182, 0.0%)** |
| Non-sustained VT | 16 (159, 10.1%) | 9 (35, 25.7%) | 7 (124, 5.6%)** |
| <50 years | 7 (119, 5.9%) | 4 (21, 19.0%) | 3 (98, 3.1%)* |
| ≥50 years | 9 (40, 22.5%) | 5 (14, 35.7%) | 4 (26, 15.4%) |
| Abnormal blood pressure response | 26 (160, 16.3%) | 7 (38, 18.4%) | 19 (122, 15.6%) |
| <50 years | 20 (117, 17.1%) | 15 (94, 16.0%) | 5 (23, 21.7%) |
| ≥50 years | 6 (43, 14.0%) | 4 (28, 14.3%) | 2 (15, 13.3%) |
| Previous cardiac arrest or VT | 1 (235, 0.4%) | 1 (53, 1.9%) | 0 (182, 0.0%) |
| Unexplained syncope | 12 (235, 5.1%) | 3 (53, 5.7%) | 9 (182, 4.9%) |
| Family history of SCD | 46 (235, 19.6%) | 6 (53, 11.3%) | 40 (182, 22.0%) |
| Number of risk factors for SCD | 0.45 ± 0.57 (235) | 0.57 ± 0.67 (53) | 0.41 ± 0.54 (182) |
| 0 risk factors | 139 (235, 59.1%) | 28 (53, 52.8%) | 111 (182, 61.0%) |
| 1 risk factors | 87 (235, 37.0%) | 20 (53, 37.7%) | 67 (182, 36.8%) |
| ≥2 risk factors | 9 (235, 3.8%) | 5 (53, 9.4%) | 4 (182, 2.2%) |
| Clinical parameters | All mutation carriers | Mutation carriers with clinical HCM | Mutation carriers without clinical HCM |
|---|---|---|---|
| Age (years) | 39.7 ± 17.9 (235) | 47.0 ± 19.0 (53) | 37.6 ± 17.1 (182)* |
| Male | 108 (235, 46.0%) | 34 (53, 64.2%) | 74 (182, 40.7%)* |
| Clinical diagnosis of HCM | 53 (235, 22.6%) | ||
| Palpitations | 49 (233, 21.0%) | 7 (51, 13.7%) | 42 (182, 23.1%) |
| Chest pain | 14 (235, 6.0%) | 5 (53, 9.4%) | 9 (182, 4.9%) |
| Atrial fibrillation | 1 (235, 0.4%) | 0 (53, 0.0%) | 1 (182, 0.5%) |
| MYBPC3 gene mutation type | |||
| c.2373_2374insG | 162 (235, 68.9%) | 42 (53, 79.2%) | 120 (182, 65.9%) |
| Other truncating mutations | 50 (235, 21.3%) | 8 (53, 15.1%) | 42 (182, 23.1%) |
| Missense mutations | 23 (235, 9.8%) | 3 (53, 5.7%) | 20 (182, 11.0%) |
| Risk factors for SCD | |||
| Extreme left-ventricular hypertrophy | 4 (235, 1.7%) | 4 (53, 7.5%) | 0 (182, 0.0%)** |
| Non-sustained VT | 16 (159, 10.1%) | 9 (35, 25.7%) | 7 (124, 5.6%)** |
| <50 years | 7 (119, 5.9%) | 4 (21, 19.0%) | 3 (98, 3.1%)* |
| ≥50 years | 9 (40, 22.5%) | 5 (14, 35.7%) | 4 (26, 15.4%) |
| Abnormal blood pressure response | 26 (160, 16.3%) | 7 (38, 18.4%) | 19 (122, 15.6%) |
| <50 years | 20 (117, 17.1%) | 15 (94, 16.0%) | 5 (23, 21.7%) |
| ≥50 years | 6 (43, 14.0%) | 4 (28, 14.3%) | 2 (15, 13.3%) |
| Previous cardiac arrest or VT | 1 (235, 0.4%) | 1 (53, 1.9%) | 0 (182, 0.0%) |
| Unexplained syncope | 12 (235, 5.1%) | 3 (53, 5.7%) | 9 (182, 4.9%) |
| Family history of SCD | 46 (235, 19.6%) | 6 (53, 11.3%) | 40 (182, 22.0%) |
| Number of risk factors for SCD | 0.45 ± 0.57 (235) | 0.57 ± 0.67 (53) | 0.41 ± 0.54 (182) |
| 0 risk factors | 139 (235, 59.1%) | 28 (53, 52.8%) | 111 (182, 61.0%) |
| 1 risk factors | 87 (235, 37.0%) | 20 (53, 37.7%) | 67 (182, 36.8%) |
| ≥2 risk factors | 9 (235, 3.8%) | 5 (53, 9.4%) | 4 (182, 2.2%) |
Data are mean ± SD or number and proportion (%).
HCM, hypertrophic cardiomyopathy; SCD, sudden cardiac death; VT, ventricular tachycardia.
Significant differences between carriers with and without a clinical diagnosis of HCM: *P-value < 0.01, **P-value < 0.001.
In 130 of 235 mutation carriers all six risk factors were evaluated. In the remaining 105 carriers on average 4.6 risk factors for SCD were evaluated. Carriers with complete evaluation were significantly younger than carriers with incomplete analysis of all risk factors (mean age 38.6 years and 41.1 years, respectively, P = 0.016). In carriers with complete evaluation a family history of SCD was more often present than in carriers with incomplete evaluation (24.6 and 13.3%, respectively, P = 0.030). There were no differences in the other clinical parameters, the proportion diagnosed with HCM and the total number of risk factors.
Men and women did not differ with respect to most clinical parameters, except that women reported palpitations more often than men (P = 0.016). A family history of SCD was less often present in females (P = 0.010).
There was no significant difference in clinical parameters between different types of MYBPC3 gene mutations (see Supplementary Data).
Diagnosis of hypertrophic cardiomyopathy
At first evaluation, a diagnosis of HCM was made in 53 of 235 asymptomatic mutation carriers (22.6%). Clinical parameters at first evaluation in mutation carriers with or without a clinical diagnosis at first visit are summarized in Table 2. Carriers who were diagnosed with HCM at first evaluation were significantly older than carriers without left-ventricular hypertrophy. Mean age of diagnosis was 47.0 years. Significantly less women than men were diagnosed with HCM (15.0 and 31.5%, respectively, P = 0.003). Mean age of diagnosis was not different in men and women; 45.5 years in men and 49.7 years in women (P = 0.437). However, disease penetrance of HCM with age was earlier in men than in women (log-rank test, P = 0.024, Figure 1). This difference between men and women holds for all types of mutations, but was not significant for each separate type of mutation.
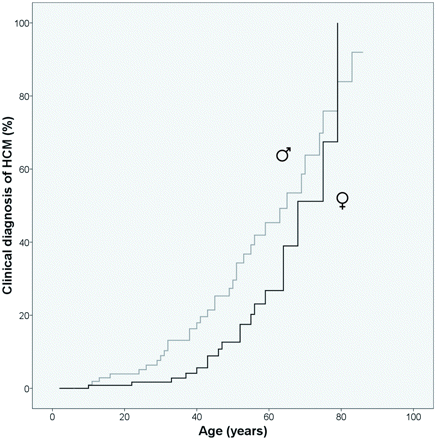
Disease penetrance of hypertrophic cardiomyopathy in male (♂) and female (♀) mutation carriers.
The proportion of mutation carriers diagnosed with HCM did not differ significantly between the different types of MYBPC3 gene mutations. Mean age of diagnosis was 46.9 (n = 42) years in carriers of the c.2373_2374insG Dutch founder mutation, 48.0 (n = 8) years in carriers of other truncating mutations, and 45.7 (n = 3) years in carriers of a missense mutation (P = 0.982). There was also no significant difference in age-related disease penetrance of HCM for different types of MYBPC3 gene mutations (Table 3).
Disease penetrance: percentage of carriers with a clinical diagnosis of hypertrophic cardiomyopathy at different ages
| Mutation type | Age (years) | |||||
|---|---|---|---|---|---|---|
| 30 | 40 | 50 | 60 | 65 | 70 | |
| c.2373insG Dutch founder mutation (n = 162), % | 4.8 | 11.8 | 23.9 | 44.2 | 52.5 | 63.1 |
| Other truncating mutations (n = 50), % | 4.6 | 7.3 | 12.4 | 17.9 | 37.4 | 37.4 |
| Missense mutations (n = 23), % | 5.9 | 13.7 | 13.7 | 13.7 | 13.7 | 42.5 |
| Mutation type | Age (years) | |||||
|---|---|---|---|---|---|---|
| 30 | 40 | 50 | 60 | 65 | 70 | |
| c.2373insG Dutch founder mutation (n = 162), % | 4.8 | 11.8 | 23.9 | 44.2 | 52.5 | 63.1 |
| Other truncating mutations (n = 50), % | 4.6 | 7.3 | 12.4 | 17.9 | 37.4 | 37.4 |
| Missense mutations (n = 23), % | 5.9 | 13.7 | 13.7 | 13.7 | 13.7 | 42.5 |
Disease penetrance: percentage of carriers with a clinical diagnosis of hypertrophic cardiomyopathy at different ages
| Mutation type | Age (years) | |||||
|---|---|---|---|---|---|---|
| 30 | 40 | 50 | 60 | 65 | 70 | |
| c.2373insG Dutch founder mutation (n = 162), % | 4.8 | 11.8 | 23.9 | 44.2 | 52.5 | 63.1 |
| Other truncating mutations (n = 50), % | 4.6 | 7.3 | 12.4 | 17.9 | 37.4 | 37.4 |
| Missense mutations (n = 23), % | 5.9 | 13.7 | 13.7 | 13.7 | 13.7 | 42.5 |
| Mutation type | Age (years) | |||||
|---|---|---|---|---|---|---|
| 30 | 40 | 50 | 60 | 65 | 70 | |
| c.2373insG Dutch founder mutation (n = 162), % | 4.8 | 11.8 | 23.9 | 44.2 | 52.5 | 63.1 |
| Other truncating mutations (n = 50), % | 4.6 | 7.3 | 12.4 | 17.9 | 37.4 | 37.4 |
| Missense mutations (n = 23), % | 5.9 | 13.7 | 13.7 | 13.7 | 13.7 | 42.5 |
Risk factors for sudden cardiac death
The cumulative number of risk factors for SCD (ranging from zero to six) was calculated for all 235 mutation carriers; 139 carriers (59%) had no risk factors, 87 carriers (37%) had one, and 9 (4%) showed two or more (Table 2, Figure 2). There was no difference in the proportion of different risk categories between men and women, between carriers of different types of mutations, between carriers of different ages, and between carriers with or without a clinical diagnosis of HCM. Twenty-five mutation carriers had a clinical diagnosis of HCM and one or more risk factors for SCD and could therefore be at high risk for SCD. Only three of them received an ICD. There were also 71 mutation carriers with one or more risk factors for SCD in whom HCM was not clinically diagnosed.
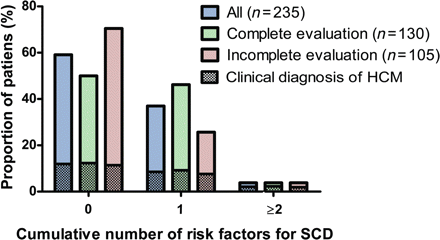
Cumulative number of risk factors for sudden cardiac death in mutation carriers.
A family history of SCD was the risk factor most frequently present. Because of the echocardiographic criteria for a diagnosis of HCM, extreme left-ventricular hypertrophy was only present in mutation carriers with a clinical diagnosis of HCM. Non-sustained ventricular tachycardia was also significantly more often present in carriers with a clinical diagnosis of HCM. This difference was only present in carriers younger than 50 years.
Complete evaluation of all six risk factors for sudden cardiac death
Not all risk factors were evaluated in all mutation carriers; particularly NSVT and ABPR were not assessed. If analysis was restricted to the 130 mutation carriers who had a complete assessment of all risk factors at first evaluation, 65 carriers (50%) had no risk factors, 60 (46%) had one risk factor, and 5 (4%) had two or more risk factors for SCD (Figure 2). In these 130 carriers there was no difference in the proportion of different risk categories between men and women, between carriers of different types of mutations, between carriers of different ages, and between carriers with or without a clinical diagnosis of HCM.
In the comparison between the 130 carriers with complete evaluation of all six risk factors and the 105 with incomplete evaluation there was no difference in the proportion diagnosed with HCM. In the group of 130 mutation carriers with complete evaluation significantly more carriers had one or more risk factor compared with the 105 mutation carriers with incomplete evaluation of risk factors (P = 0.005). There was no difference in maximal left-ventricular wall thickness between the carriers in whom all risk factors had been assessed and carriers who had been incompletely evaluated. From the evaluated risk factors only the presence of a family history of SCD was significantly different. This risk factor was more often present in carriers with complete evaluation than in carriers with incomplete evaluation (P = 0.030).
Follow-up
During a mean follow-up of 3.2 years two mutation carriers died. One was a female who was diagnosed with HCM at first evaluation. Five years after her first evaluation she died at the age of 80 years after an unsuccessful resuscitation from ventricular fibrillation. Autopsy showed marked hypertrophy of the left ventricle. The other mutation carrier died at the age of 50 years from metastasized colon cancer. She died more than 3 years after her first cardiological evaluation which showed no signs of HCM. Both mutation carriers had no risk factors for SCD.
Discussion
This is to the best of our knowledge the first study assessing not only the presence of left-ventricular hypertrophy but also risk factors for SCD in asymptomatic carriers of MYBPC3 mutations identified through cascade screening. In our group of 235 carriers HCM was diagnosed in almost one-quarter at their first evaluation. Mean age of diagnosis was higher than in studies of HCM patients previously reported in the literature, probably because mutation carriers screened in a predictive setting represent an asymptomatic subgroup within the total population of affected HCM patients.14,17–20
Besides age, gender was an apparent cofactor in the clinical manifestation of HCM. At first evaluation a clinical diagnosis was less often made in female than in male mutation carriers. Male and female mutation carriers did not differ with respect to age at first visit. Although the mean age of diagnosis between males and females is not significantly different, the Kaplan–Meier curve, which also takes into account the mutation carriers in whom a clinical diagnosis could not be made and their age at that time, showed that women were diagnosed at an higher age than men. This is in agreement with the literature, which shows an overrepresentation of men with HCM in almost all studies and females diagnosed at an older age.14,20
In patients with the c.2373_2374insG Dutch founder mutation the clinical diagnosis of HCM was just as often made as in other types of MYBPC3 mutations and at the same age, showing that the c.2373_2374insG mutation is just as potent in causing HCM as other MYBPC3 gene mutations. This is in line with the suggested pathogenetic mechanism of MYBPC3 mutations, being total or partial haploinsufficiency.21 Kaplan–Meier curves demonstrated that disease penetrance was age dependent and far from complete in all types of MYBPC3 mutations at the age of 65 years. The majority of mutation carriers were even diagnosed with HCM after the age of 50 years. These data contrast with the literature on MYBPC3 mutations, showing that disease penetrance for MYBPC3 mutations is incomplete but highly penetrant,22,23 with disease penetrance over 40% at the age of 30 (around 5% in our mutation carriers), and in mutation carriers older than 60 years around 90% (<50% in our mutation carriers).23 Our data suggest that cardiological evaluations should be continued until advanced age in mutation carriers, irrespective of the type of MYBPC3 mutation.
Data on the prevalence of risk factors for SCD in asymptomatic mutation carriers have not been described earlier. We showed that these risk factors appeared to be frequently present in this population and the cumulative number of risk factors was not related to age, sex, mutation type, and presence of left-ventricular hypertrophy. Because NSVT and ABPR were not measured in all mutation carriers—as appears to be daily practice all over the world9,17,19 the total number of risk factors was presented for the 130 mutation carriers in whom all six risk factors were evaluated and for the 105 carriers with incomplete evaluation. This showed a significantly lower prevalence of one or more risk factors for SCD in the latter group, which suggests an underestimation of the cumulative number of risk factors due to incomplete evaluation.
In the literature, the presence of one or more risk factors for SCD in HCM patients (i.e. with a clinical diagnosis of HCM) is significantly associated with an increased risk of sudden death compared with HCM patients with no risk factors.7,8,10 In 25 of 96 mutation carriers with one or more risk factors (11% of the entire group and 47% of carriers diagnosed with HCM), although asymptomatic, a diagnosis of HCM was made and they could be at high risk for SCD. Recently, Maron et al.9 described that in HCM patients with an ICD the likelihood of appropriate discharge was similar in patients with one, two, three, or more risk factors. These data suggest that the risk for SCD is as high in patients with one, two, or more risk factors, although risk stratification in the above-mentioned study was incomplete not taking into account ABPR and with NSVT not being evaluated in almost 20% of patients. Current guidelines advocate that ICD implantation can be effective and is reasonable for the prevention of SCD in HCM patients with one or more risk factors.8,10 If these guidelines would be implemented straightforward, at least 25 (11%) asymptomatic patients identified through cascade screening should receive an ICD directly after their first cardiological visit. We feel that in HCM patients—and mutation carriers with a clinical diagnosis of HCM—with only one risk factor, the decision to implant an ICD should be made on a case-by-case basis because of the limited positive predictive value of a single risk factor.
In mutation carriers without a diagnosis of HCM, NSVT was present in 5.6% and an ABPR in 15.6%. In asymptomatic apparently healthy individuals NSVT is present in 0–3%.24 It is known that NSVT can be the first manifestation of cardiomyopathy.25 An ABPR has been shown in 2% of patients from a university medical centre.26 These two risk factors seem to occur more frequently in our unaffected mutation carriers than in healthy subjects and are therefore likely to be related to HCM, although HCM was clinically not (yet) detectable.
In follow-up only one SCD was recorded in a mutation carrier with a clinical diagnosis of HCM but without risk factors for SCD. No SCD or HCM-related death occurred in mutation carriers without a diagnosis of HCM during a follow-up of 3 years. In both subgroups no conclusions can be drawn about the prognostic impact of risk factors. The prognostic significance of risk factors for SCD needs to be further evaluated in HCM mutation carriers and especially in carriers without left-ventricular hypertrophy.
The definition used for a positive family history of SCD is the most stringent and most used definition in literature. None of the definitions used in the literature, including the one we used, takes into account the size of the family or the death of a young child. If we would have used a different definition for a positive family history of SCD in our cohort, the total number of mutation carriers with this risk factors would not differ, but in the individual carrier the total number of risk factors would vary according to the used definition of a family history of SCD. The prognostic impact of a family history for premature SCD in HCM patients has to be defined in future studies using different definitions for a family history of SCD in the same study.
Study limitations
Although our group of mutation carriers was of considerable size, distribution of a few characteristics was skewed. Significantly less men than women were included. One possible explanation is that more female relatives attend for predictive genetic counselling and testing. Results may be biased because of this, although only few clinical parameters were significantly different between men and women.
Most carriers had the c.2373_2374insG Dutch founder mutation. Although there was no association between the type of mutation and most clinical parameters, definite genotype–phenotype correlations with respect to age of diagnosis and risk factors for SCD cannot be made. Because we only included carriers of an MYBPC3 gene mutation, our conclusions cannot be generalized to carriers of mutations in other genes associated with HCM. Mutations in the MYBPC3 gene, however, are worldwide one of the most frequent causes of HCM, accounting for 15–30% of all identified HCM mutations.11,21,27
Because we referred mutation carriers to a local hospital in the proximity of their home not all mutation carriers received a Holter recording and/or exercise test at first cardiological evaluation. Other studies also show that complete risk stratification is not a customary practice in HCM patients, not to mention in asymptomatic HCM mutation carriers.9,17,19 We ruled out the possibility of absence of a clinical diagnosis of HCM being a cause for an incomplete cardiological evaluation. There was also no association between other clinical parameters and the completeness of the cardiological evaluation, except the presence of a family history of SCD, giving us no reason to believe that the evaluation of a risk factor was systematically not performed. Because one or more risk factors were less often present in the carriers without a complete evaluation of risk factors, we even think that the cumulative number of risk factors for SCD in the entire cohort is an underestimation.
Predictive genetic screening occurred in tertiary care centres. Because this is the only setting where genetic testing for HCM is possible in the Netherlands, we do not expect selection bias based upon centre.
Because asymptomatic relatives were only sent to a cardiologist after the detection of the familial mutation and because affected relatives and probands were excluded, the age-related disease penetrance does not reflect the natural history of HCM. Detection of clinical HCM in this study is also related to the age people were tested. It is possible that those with a clinical diagnosis of HCM had HCM at an earlier age but the diagnosis was not made until a later age when the DNA testing became available.
At present follow-up is too short and the number of SCDs is too small to draw meaningful conclusions on the prognostic significance of risk factors for SCD in this group of asymptomatic mutation carriers. However, we do intend to continue follow-up in this cohort to learn more about risk stratification especially in HCM mutation carriers without left-ventricular hypertrophy.
Conclusions
We can conclude that in a substantial proportion of the asymptomatic mutation carriers of an MYBPC3 gene mutation, identified through cascade screening, a diagnosis of HCM can be made at the first cardiological evaluation. Disease penetrance, however, is far from complete until advanced age and women are diagnosed at an older age than men. Risk factors for SCD are frequently present and 11% of asymptomatic mutation carriers could be at risk for SCD (a diagnosis of HCM and the presence of one or more risk factors). Risk factors are also present in mutation carriers without a clinical diagnosis of HCM, but longer follow-up studies are needed to assess if these risk factors are also associated with an increased risk of SCD in carriers without a clinical diagnosis of HCM. The type of MYBPC3 gene mutation does not seem to have a major role in clinical expression.
Our data justify predictive DNA testing in HCM families with a pathogenic mutation, regular cardiological evaluations on the presence of HCM and risk factors for SCD until advanced age.
Supplementary material
Funding
This work was supported by ZonMw [grant number 62000010]; and the Netherlands Heart Foundation [grant number 2003 D302]. The funding organizations have had no involvement in study design, collection, analysis, and interpretation of data; in the writing of this paper, and in the decision to submit the paper for publication.
Conflict of interest: none declared.


{kind=link}
{kind=link}