





Abstract
Direct renin inhibitors provide an alternative approach to inhibiting the renin–angiotensin–aldosterone system (RAAS) at the most proximal, specific, and rate-limiting step. We tested the hypothesis that direct renin inhibition would attenuate left ventricular remodelling in patients following acute myocardial infarction receiving stable, individually optimized therapy, including another inhibitor of the RAAS.
We randomly assigned 820 patients between ∼2 and 8 weeks following acute myocardial infarction, with the left ventricular ejection fraction (LVEF) ≤45%, and regional wall motion abnormalities (≥20% akinetic area), to receive aliskiren (n = 423), titrated to 300 mg, or matched placebo (n = 397), added to the standard therapy. All patients were required to be on a stable dose of an ACE-inhibitor or ARB, and beta-blocker unless contraindicated or not tolerated. Echocardiograms were obtained at baseline, and following 26–36 weeks of treatment. The primary endpoint was change in left ventricular end-systolic volume from baseline to 36 weeks, and was evaluable in 329 patients in the placebo group and 343 patients in the aliskiren group. We observed no difference in the primary endpoint of end-systolic volume change between patients randomized to aliskiren (−4.4 ± 16.8 mL) or placebo (−3.5 ± 16.3 mL), or in secondary measures of end-diastolic volume, or LVEF. We also observed no differences in a composite endpoint of cardiovascular death, hospitalization for heart failure, or reduction in LVEF >6 points. There were more investigator reported adverse events in the aliskiren group, including hypotension, increases in creatinine and hyperkalaemia.
Adding the direct renin inhibitor aliskiren to the standard therapy, including an inhibitor of the RAAS, in high-risk post-MI patients did not result in further attenuation of left ventricular remodelling, and was associated with more adverse effects. These findings do not suggest that dual RAAS blockade with aliskiren would provide additional benefit in these high-risk post-MI patients. Clinical Trials Registration: www.clinicaltrials.gov NCT00414609
Introduction
Morbidity and mortality following acute myocardial infarction (AMI) remain high despite major therapeutic advances. In post-MI patients, reduced left ventricular (LV) systolic function is associated with increased risk of LV remodelling, heart failure, and mortality. Angiotensin-converting enzyme inhibitors (ACE-I) decrease these risks1–3 and angiotensin receptor blockers (ARBs) can be an effective alternative to ACE-I in this population. Combining these two classes of RAAS inhibitors, however, in the Valsartan in Acute Myocardial Infarction (VALIANT) trial4 did not show incremental advantage in post-infarction patients, yet was associated with more side-effects, in contrast to findings in patients with chronic heart failure where some additional benefit from combination therapy was seen in two separate trials.5,6 Possible explanations for this discrepancy include the fact that most of the patients in VALIANT were ACE-I naïve when started on two RAAS inhibitors simultaneously in the acute phase of infarction, whereas in the heart failure trials the second inhibitor was added in patients chronically treated with another blocker usually for months, if not years.
As the excess of hypotension and renal dysfunction with combination therapy in VALIANT suggests that monotherapy had not fully blocked the RAAS, we hypothesized that adding a full dose of a second inhibitor of the RAAS post-MI would have beneficial effects, as in chronic heart failure, if started in the sub-acute phase, where combination therapy might be better tolerated and sustained. This hypothesis was supported by the post hoc finding in VALIANT of fewer hospitalizations for heart failure and MI with captopril and valsartan compared with captopril alone4.
The present study was specifically designed to test the hypothesis that adding the direct renin-inhibitor aliskiren to standard therapy, including a stable ACE-I or ARB dose, would result in greater attenuation of adverse LV remodelling after AMI. Aliskiren blocks the renin–angiotensin–aldosterone system proximally at the rate-limiting step,7,8 preventing the compensatory rise in plasma renin activity and other downstream components of this system which occurs in the setting of ACE-I or ARB therapy. These compensatory changes may lead to reactivation of the RAAS and it has also been suggested that plasma renin activity itself has potentially detrimental actions. Prior mechanistic studies have shown favourable effects of adding aliskiren to another inhibitor of the RAAS in patients with hypertension,9,10 heart failure,11 or diabetic kidney disease.12 The study was designed as a proof of concept trial to determine whether this therapy should be more fully evaluated in a subsequent morbidity and mortality trial.
Methods
Patients
We studied stable patients at high risk of LV remodelling after AMI. Patients were eligible for enrolment if they were over 18 years old, were between 2 and 8 weeks after AMI, and were on stable doses for 2 weeks of an antiplatelet agent, a statin, a beta-blocker and a physician determined optimal dose of either an ACE-I or ARB.13 Moreover, to be eligible patients underwent a screening echocardiogram on which they were required to have an ejection fraction ≤45% and infarct size ≥20%, as assessed in a core laboratory (see below). Patients receiving both an ACE-I and ARB were excluded, as were those with refractory hypertension, eGFR <30 mL/min/1.72 m2 (measured by the MDRD formula), or serum potassium >5 mmol/L (see Supplementary Data for complete listing of inclusion and exclusion criteria). Aldosterone antagonists were allowed at the discretion of the treating physicians in view of the beneficial effect of this type of treatment in patients with a reduced LVEF and heart failure after myocardial infarction.14
ASPIRE was conducted in 154 centres from 23 countries. We screened 1074 consenting patients for potential inclusion in the study (Figure 1). Of these, 820 fulfilled inclusion criteria and 397 patients were randomized to the placebo group and 423 patients to the aliskiren group. A total of 422 patients actually received aliskiren. Sixty-eight patients in the placebo group and 80 patients in the aliskiren group either died, withdrew consent or had echocardiograms that were of insufficient quality to be included in the evaluation, leaving 329 patients in the placebo group and 343 patients in the aliskiren group with evaluable echocardiograms available at baseline and at end of the study.
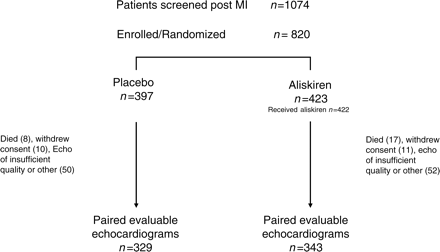
Patient flow.
Patients who qualified by echocardiographic criteria were randomized to aliskiren or placebo, beginning at a dose of 75 mg and titrated up to 300 mg once daily for 36 weeks. Randomization numbers were generated using a validated IVRS system that automated the random assignment of patient numbers to randomization numbers in an unbiased fashion and concealed randomization codes from patients and investigator staff. Patients, investigator staff, persons performing the assessments, and data analysts remained blinded to the identity of the treatment from the time of randomization until database lock. Randomization data was kept strictly confidential until the time of unblinding, and was not accessible by anyone else involved in the study with the exception of authorized persons, and the identity of the treatments was concealed by the use of study drugs that were identical in packaging, labelling, schedule of administration, and appearance. A double-dummy design was used because the identity of the study drugs could not be disguised due to their different forms. Randomization was stratified according to whether a patient was taking an aldosterone blocker at baseline and by centre. Patients were followed for 36 weeks after which the final echocardiogram was obtained. Details of the study design and titration scheme are shown in Figure 2.
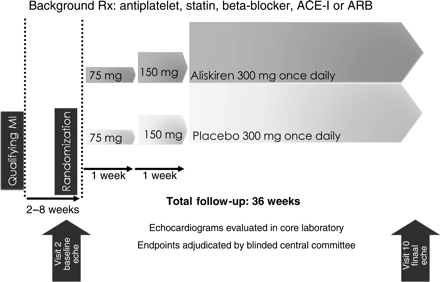
Study design and titration scheme.
Efficacy measures
The primary endpoint of the trial was change in end-systolic volume from baseline to 36 weeks. Key secondary endpoints included a composite of CV death, hospitalization for heart failure, or a reduction in ejection fraction >6 units and a composite of CV death, hospitalization for heart failure, recurrent myocardial infarction, stroke, or resuscitated sudden death. Additionally, we assessed the overall safety and tolerability of aliskiren in combination with the standard therapy and assessed other echocardiographic assessments of cardiac size and function.
Echocardiographic measurements
Site sonographers received training in optimal echocardiographic assessment for determining ventricular volumes at investigators meetings and site certification was performed. Echocardiograms were transferred on CD or videotape to a core laboratory at Brigham and Women's Hospital. Ventricular volumes were determined by the modified Simpson's method in the apical four and two chamber views, and the ejection fraction was calculated from volumes in the standard manner.15 Infarct segment length was assessed as length of the akinetic or dyskinetic endocardium as a percentage of the total perimeter in each view. All echocardiographic images were assessed for image quality and for foreshortening that could adversely affect volumetric measurements. Studies with poor image quality or in which substantial foreshortening was observed were excluded from the analyses.
Clinical outcomes and adverse events
Major adverse cardiovascular events, including death, recurrent myocardial infarction, stroke, and heart failure hospitalization, were adjudicated by a central endpoint committee blinded to treatment assignment according to prespecified standard definitions.
All site investigators reported adverse events and serious adverse events that were potential endpoints were submitted for adjudication. Adverse and serious adverse events that represented study endpoints were reported separately from investigator reported adverse events.
Statistical considerations
For the primary endpoint (change in LVESV from baseline to week 36), the study was designed to have 80% power to detect a 3.1 mL difference in change in LVESV at a two-sided 0.05 significance level, with an estimated total sample size of 800 patients. The prespecified primary analysis was conducted using the analysis of covariance adjusting for stratification variables, including region, use of aldosterone antagonists, as well as baseline LVESV and was based on intent to treat in patients with both baseline and follow-up echocardiographic assessment. Non-echocardiographic efficacy endpoints were reported on a strict intent to treat basis. Adverse events and safety lab measurements were reported for treated patients.
Baseline characteristics between groups were compared with Student's t-test for continuous variables and χ2 test for categorical variables. Time-to-event analyses treatment comparisons were made using Cox regression, adjusted for age, history of prior MI, and baseline LVESV. Adverse event and safety lab comparisons were made using Fisher's exact test. Post hoc subgroup analyses were performed in the following subgroups: age ≥ or <65, gender, history of diabetes mellitus, history of hypertension, use of aldosterone antagonists, and left ventricular ejection fraction ≤35%. Interaction was assessed between subgroups and the primary outcome, adjusting for baseline end-systolic volume and stratification variables as with the analysis of the primary endpoint. A P-value of < 0.05 was considered significant, and all between treatment group comparisons were two-sided. All analyses were performed using Stata, version 11, at Brigham and Women's Hospital and confirmed using SAS, version 9.2, by the sponsor.
The study was designed jointly by the academic steering committee and the sponsor. The sponsor was involved in study management, data collection, and data analysis. At the completion of the trial, all data were transferred to Brigham and Women's Hospital and analysed by an independent academic statistician. The manuscript was written by the academic steering committee. The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree with the manuscript as written.
Results
Baseline characteristics
Baseline characteristics of the patients randomized are shown in Table 1. On average, patients were ∼60 years old, primarily male, and had a baseline eGFR of 80 mL/min/1.72 m2. The majority of patients had Q-wave, anterior location infarctions. There were no statistically significant differences between groups, although a greater percentage of patients randomized to the aliskiren group were over 65 years old, had eGFR <60 mL/min/1.72 m2, had a history of prior MI or heart failure, and were taking aldosterone antagonists. Almost all were receiving antiplatelet agents, statins, beta-blockers, and ACE-Is or ARB at baseline. Forty-four per cent of patients were taking an optimal dose of an ACE-I or ARB, defined as daily doses of 150 mg of captopril, 20 mg of enalapril, 20 mg of lisinopril, 8 mg of perindopril, 10 mg of ramipril, 32 mg of candesartan, 320 mg of valsartan, 100 mg of losartan, or 300 mg of irbesartan. Approximately 27% of patients were taking an aldosterone blocker. The baseline left ventricular end-diastolic volume in all patients who had baseline data available was ∼132 mL and end-systolic volume ∼84 mL. The average ejection fraction was 37.5%, and the infarct segment length was just over 25%. Baseline blood pressures were 121.6 ± 16.1/75.2 ± 9.4 in the aliskiren group and 121.7 ± 16.2/75.4 ± 9.3 in the placebo group. The median time to randomization following MI was 43 days.
Baseline characteristics
| Characteristic | Placebo, n = 397 | Aliskiren, n = 423 |
|---|---|---|
| Male gender (%) | 85 | 81 |
| Age (years ± SD) | 59 ± 12 | 61 ± 12 |
| Age ≥65 years (%) | 34 | 40 |
| Baseline eGFR | 81 ± 19 | 80 ± 21 |
| (mL/min/1.73 m2) (%) | ||
| eGFR<60 | 13 | 17 |
| History of diabetes | 22 | 23 |
| History of hypertension | 50 | 55 |
| History of MI | 18 | 22 |
| Prior HF hospitalization | 4 | 6 |
| Infarct type (%) | ||
| Q-wave MI | 71 | 68 |
| Anterior MI | 79 | 79 |
| Killip class ≥2 | 42 | 45 |
| Reperfusion Therapy | 76 | 73 |
| Baseline medications (%) | ||
| Anti-platelet agents | 98 | 98 |
| ACE-I | 91 | 89 |
| ARB | 9 | 10 |
| ‘Optimal’ dose ACE-I or ARB | 43 | 44 |
| Beta-blocker | 95 | 96 |
| Statin | 98 | 97 |
| Aldosterone blocker | 24 | 29 |
| Baseline echocardiographic measures | ||
| LVESV (mL) | 86.1 ± 29.9 | 82.4 ± 26.0 |
| LVEDV (mL) | 135.7 ± 36.2 | 130.5 ± 32.9 |
| Left ventricular ejection fraction (%) | 37.5 ± 5.8 | 37.6 ± 5.3 |
| Infarct length (%) | 25.0 ± 10.6 | 25.7 ± 10.6 |
| Characteristic | Placebo, n = 397 | Aliskiren, n = 423 |
|---|---|---|
| Male gender (%) | 85 | 81 |
| Age (years ± SD) | 59 ± 12 | 61 ± 12 |
| Age ≥65 years (%) | 34 | 40 |
| Baseline eGFR | 81 ± 19 | 80 ± 21 |
| (mL/min/1.73 m2) (%) | ||
| eGFR<60 | 13 | 17 |
| History of diabetes | 22 | 23 |
| History of hypertension | 50 | 55 |
| History of MI | 18 | 22 |
| Prior HF hospitalization | 4 | 6 |
| Infarct type (%) | ||
| Q-wave MI | 71 | 68 |
| Anterior MI | 79 | 79 |
| Killip class ≥2 | 42 | 45 |
| Reperfusion Therapy | 76 | 73 |
| Baseline medications (%) | ||
| Anti-platelet agents | 98 | 98 |
| ACE-I | 91 | 89 |
| ARB | 9 | 10 |
| ‘Optimal’ dose ACE-I or ARB | 43 | 44 |
| Beta-blocker | 95 | 96 |
| Statin | 98 | 97 |
| Aldosterone blocker | 24 | 29 |
| Baseline echocardiographic measures | ||
| LVESV (mL) | 86.1 ± 29.9 | 82.4 ± 26.0 |
| LVEDV (mL) | 135.7 ± 36.2 | 130.5 ± 32.9 |
| Left ventricular ejection fraction (%) | 37.5 ± 5.8 | 37.6 ± 5.3 |
| Infarct length (%) | 25.0 ± 10.6 | 25.7 ± 10.6 |
Baseline characteristics
| Characteristic | Placebo, n = 397 | Aliskiren, n = 423 |
|---|---|---|
| Male gender (%) | 85 | 81 |
| Age (years ± SD) | 59 ± 12 | 61 ± 12 |
| Age ≥65 years (%) | 34 | 40 |
| Baseline eGFR | 81 ± 19 | 80 ± 21 |
| (mL/min/1.73 m2) (%) | ||
| eGFR<60 | 13 | 17 |
| History of diabetes | 22 | 23 |
| History of hypertension | 50 | 55 |
| History of MI | 18 | 22 |
| Prior HF hospitalization | 4 | 6 |
| Infarct type (%) | ||
| Q-wave MI | 71 | 68 |
| Anterior MI | 79 | 79 |
| Killip class ≥2 | 42 | 45 |
| Reperfusion Therapy | 76 | 73 |
| Baseline medications (%) | ||
| Anti-platelet agents | 98 | 98 |
| ACE-I | 91 | 89 |
| ARB | 9 | 10 |
| ‘Optimal’ dose ACE-I or ARB | 43 | 44 |
| Beta-blocker | 95 | 96 |
| Statin | 98 | 97 |
| Aldosterone blocker | 24 | 29 |
| Baseline echocardiographic measures | ||
| LVESV (mL) | 86.1 ± 29.9 | 82.4 ± 26.0 |
| LVEDV (mL) | 135.7 ± 36.2 | 130.5 ± 32.9 |
| Left ventricular ejection fraction (%) | 37.5 ± 5.8 | 37.6 ± 5.3 |
| Infarct length (%) | 25.0 ± 10.6 | 25.7 ± 10.6 |
| Characteristic | Placebo, n = 397 | Aliskiren, n = 423 |
|---|---|---|
| Male gender (%) | 85 | 81 |
| Age (years ± SD) | 59 ± 12 | 61 ± 12 |
| Age ≥65 years (%) | 34 | 40 |
| Baseline eGFR | 81 ± 19 | 80 ± 21 |
| (mL/min/1.73 m2) (%) | ||
| eGFR<60 | 13 | 17 |
| History of diabetes | 22 | 23 |
| History of hypertension | 50 | 55 |
| History of MI | 18 | 22 |
| Prior HF hospitalization | 4 | 6 |
| Infarct type (%) | ||
| Q-wave MI | 71 | 68 |
| Anterior MI | 79 | 79 |
| Killip class ≥2 | 42 | 45 |
| Reperfusion Therapy | 76 | 73 |
| Baseline medications (%) | ||
| Anti-platelet agents | 98 | 98 |
| ACE-I | 91 | 89 |
| ARB | 9 | 10 |
| ‘Optimal’ dose ACE-I or ARB | 43 | 44 |
| Beta-blocker | 95 | 96 |
| Statin | 98 | 97 |
| Aldosterone blocker | 24 | 29 |
| Baseline echocardiographic measures | ||
| LVESV (mL) | 86.1 ± 29.9 | 82.4 ± 26.0 |
| LVEDV (mL) | 135.7 ± 36.2 | 130.5 ± 32.9 |
| Left ventricular ejection fraction (%) | 37.5 ± 5.8 | 37.6 ± 5.3 |
| Infarct length (%) | 25.0 ± 10.6 | 25.7 ± 10.6 |
Blood pressure
The mean sitting blood pressure throughout the trial was slightly lower in the aliskiren group compared with placebo, with final blood pressures of 122.1 ± 16.0/74.0 ± 9.3 in the aliskiren group and 124.2 ± 14.9/76.4 ± 9.4 in the placebo group (between-treatment change: systolic BP change, P = 0.09; diastolic BP change, P = 0.001).
Efficacy measures
End-systolic volume decreased by 3.5 ± 16.3 mL in the placebo group and by 4.4 ± 16.8 mL in aliskiren group (Table 2; P = 0.44). Similarly, the between group changes in end-diastolic volume, ejection fraction, infarct segment length, or percent of patients whose ejection fraction decreased by >6 points did not differ significantly (Table 2). There were no differences in any of the clinical secondary efficacy variables (Table 3), including the composite of cardiovascular death, hospitalization for heart failure or reduction in LVEF by >6 points, or the clinical composite of cardiovascular death, hospitalization for heart failure, recurrent MI, stroke, or resuscitated sudden death. All-cause mortality was only 2% in the placebo group and numerically, but not statistically, higher in the aliskiren group (Table 3). A similar proportion of deaths (6/8 in the placebo group and 13/17 in the aliskiren group) were adjudicated as cardiovascular. There were no significant differences between treatment groups in the components of the adjudicated endpoints. Within post hoc subgroups analysed, there were no differences in the treatment effect, with the exception of the diabetic subgroup, for whom we observed a borderline interaction in favour of aliskiren (Figure 3).
Changes in echocardiographic measures
| Placebo, n = 329 | Aliskiren, n = 343 | P-value | |||
|---|---|---|---|---|---|
| Baseline | Change | Baseline | Change | ||
| LVESV(mL) | 84.2 ± 25.5 | −3.5 ± 16.3 | 82.5 ± 26.6 | −4.4 ± 16.8 | 0.44 |
| LVEDV (mL) | 133.5 ± 31.6 | −1.7 ± 19.6 | 131.2 ± 33.9 | −3.2 ± 19.3 | 0.26 |
| LVEF (%) | 37.7 ± 5.5 | 2.3 ± 4.1 | 37.9 ± 5.1 | 2.5 ± 4.5 | 0.71 |
| Infarct | |||||
| Length (%) | 24.4 ± 10.3 | −4.7 ± 9.5 | 25.1 ± 10.4 | −5.6 ± 9.0 | 0.27 |
| EF drop > 6% | 2 (0.6%) | 6 (1.8%) | 0.17 | ||
| Placebo, n = 329 | Aliskiren, n = 343 | P-value | |||
|---|---|---|---|---|---|
| Baseline | Change | Baseline | Change | ||
| LVESV(mL) | 84.2 ± 25.5 | −3.5 ± 16.3 | 82.5 ± 26.6 | −4.4 ± 16.8 | 0.44 |
| LVEDV (mL) | 133.5 ± 31.6 | −1.7 ± 19.6 | 131.2 ± 33.9 | −3.2 ± 19.3 | 0.26 |
| LVEF (%) | 37.7 ± 5.5 | 2.3 ± 4.1 | 37.9 ± 5.1 | 2.5 ± 4.5 | 0.71 |
| Infarct | |||||
| Length (%) | 24.4 ± 10.3 | −4.7 ± 9.5 | 25.1 ± 10.4 | −5.6 ± 9.0 | 0.27 |
| EF drop > 6% | 2 (0.6%) | 6 (1.8%) | 0.17 | ||
Treatment group values shown are mean ± standard deviation and n (%).
P-value for EF drop >6% based on χ2 test.
P-values for LVESV, LVEDV, LVEF, and infarct length are from analyses of covariance of change from baseline, adjusted for region, aldosterone antagonist use, and baseline measurement.
Changes in echocardiographic measures
| Placebo, n = 329 | Aliskiren, n = 343 | P-value | |||
|---|---|---|---|---|---|
| Baseline | Change | Baseline | Change | ||
| LVESV(mL) | 84.2 ± 25.5 | −3.5 ± 16.3 | 82.5 ± 26.6 | −4.4 ± 16.8 | 0.44 |
| LVEDV (mL) | 133.5 ± 31.6 | −1.7 ± 19.6 | 131.2 ± 33.9 | −3.2 ± 19.3 | 0.26 |
| LVEF (%) | 37.7 ± 5.5 | 2.3 ± 4.1 | 37.9 ± 5.1 | 2.5 ± 4.5 | 0.71 |
| Infarct | |||||
| Length (%) | 24.4 ± 10.3 | −4.7 ± 9.5 | 25.1 ± 10.4 | −5.6 ± 9.0 | 0.27 |
| EF drop > 6% | 2 (0.6%) | 6 (1.8%) | 0.17 | ||
| Placebo, n = 329 | Aliskiren, n = 343 | P-value | |||
|---|---|---|---|---|---|
| Baseline | Change | Baseline | Change | ||
| LVESV(mL) | 84.2 ± 25.5 | −3.5 ± 16.3 | 82.5 ± 26.6 | −4.4 ± 16.8 | 0.44 |
| LVEDV (mL) | 133.5 ± 31.6 | −1.7 ± 19.6 | 131.2 ± 33.9 | −3.2 ± 19.3 | 0.26 |
| LVEF (%) | 37.7 ± 5.5 | 2.3 ± 4.1 | 37.9 ± 5.1 | 2.5 ± 4.5 | 0.71 |
| Infarct | |||||
| Length (%) | 24.4 ± 10.3 | −4.7 ± 9.5 | 25.1 ± 10.4 | −5.6 ± 9.0 | 0.27 |
| EF drop > 6% | 2 (0.6%) | 6 (1.8%) | 0.17 | ||
Treatment group values shown are mean ± standard deviation and n (%).
P-value for EF drop >6% based on χ2 test.
P-values for LVESV, LVEDV, LVEF, and infarct length are from analyses of covariance of change from baseline, adjusted for region, aldosterone antagonist use, and baseline measurement.
Outcome measures
| Placebo, n = 397 (%) | Aliskiren, n = 423 (%) | HR | 95% CI | P-value | |
|---|---|---|---|---|---|
| Composite of CV death, hospitalization for HF, LVEF reduction by >6 units | 24 (6) | 29 (7) | 1.06 | (0.60, 1.85) | 0.85 |
| Composite of CV death, hospitalization for HF, recurrent MI, stroke, resuscitated sudden death | 34 (9) | 39 (9) | 1.01 | (0.62, 1.63) | 0.98 |
| All-cause death | 7 (2) | 15 (4) | 1.77 | (0.72, 4.36) | 0.22 |
| All-cause death without baseline LVESV covariate | 8 (2) | 17 (4) | 1.76 | (0.76, 4.10) | 0.19 |
| Placebo, n = 397 (%) | Aliskiren, n = 423 (%) | HR | 95% CI | P-value | |
|---|---|---|---|---|---|
| Composite of CV death, hospitalization for HF, LVEF reduction by >6 units | 24 (6) | 29 (7) | 1.06 | (0.60, 1.85) | 0.85 |
| Composite of CV death, hospitalization for HF, recurrent MI, stroke, resuscitated sudden death | 34 (9) | 39 (9) | 1.01 | (0.62, 1.63) | 0.98 |
| All-cause death | 7 (2) | 15 (4) | 1.77 | (0.72, 4.36) | 0.22 |
| All-cause death without baseline LVESV covariate | 8 (2) | 17 (4) | 1.76 | (0.76, 4.10) | 0.19 |
Hazard ratios (HR), 95% HR confidence intervals (CI), and P-values are from a Cox regression adjusted for age, prior MI, and baseline LVESV unless otherwise noted.
Outcome measures
| Placebo, n = 397 (%) | Aliskiren, n = 423 (%) | HR | 95% CI | P-value | |
|---|---|---|---|---|---|
| Composite of CV death, hospitalization for HF, LVEF reduction by >6 units | 24 (6) | 29 (7) | 1.06 | (0.60, 1.85) | 0.85 |
| Composite of CV death, hospitalization for HF, recurrent MI, stroke, resuscitated sudden death | 34 (9) | 39 (9) | 1.01 | (0.62, 1.63) | 0.98 |
| All-cause death | 7 (2) | 15 (4) | 1.77 | (0.72, 4.36) | 0.22 |
| All-cause death without baseline LVESV covariate | 8 (2) | 17 (4) | 1.76 | (0.76, 4.10) | 0.19 |
| Placebo, n = 397 (%) | Aliskiren, n = 423 (%) | HR | 95% CI | P-value | |
|---|---|---|---|---|---|
| Composite of CV death, hospitalization for HF, LVEF reduction by >6 units | 24 (6) | 29 (7) | 1.06 | (0.60, 1.85) | 0.85 |
| Composite of CV death, hospitalization for HF, recurrent MI, stroke, resuscitated sudden death | 34 (9) | 39 (9) | 1.01 | (0.62, 1.63) | 0.98 |
| All-cause death | 7 (2) | 15 (4) | 1.77 | (0.72, 4.36) | 0.22 |
| All-cause death without baseline LVESV covariate | 8 (2) | 17 (4) | 1.76 | (0.76, 4.10) | 0.19 |
Hazard ratios (HR), 95% HR confidence intervals (CI), and P-values are from a Cox regression adjusted for age, prior MI, and baseline LVESV unless otherwise noted.
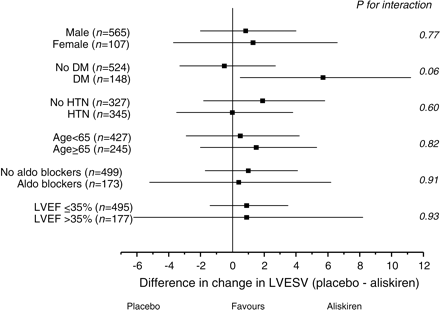
Post hoc subgroup analysis. Aldo, aldosterone.
Adverse events
Overall there were more patient or investigator reported adverse events in the patients receiving aliskiren (P = 0.02; Table 4). However, the total number of serious adverse events was similar in the two arms (P = 0.51). In particular, there were more investigator reported renal dysfunction (P = 0.09), hypotension (P = 0.02), and hyperkalaemia events (P = 0.001) in the aliskiren group, but serious adverse events were similar within these categories as well. Elevations in blood urea nitrogen and creatinine were more likely in the aliskiren group, and patients in the aliskiren group were more likely to have a potassium value measured at >5.5 or >6.0 mmol/L (Table 5).
Investigator and patient reported adverse events
| Placebo (n = 397) | Aliskiren (n = 422a) | P-value | |
|---|---|---|---|
| Total adverse events (AEs) | 268 (67.5) | 316 (74.9) | 0.02 |
| Total serious adverse events (SAEs) | 92 (23.2) | 107 (25.4) | 0.51 |
| Renal dysfunction | |||
| AEs | 3 (0.8) | 10 (2.4) | 0.09 |
| SAEs | 1 (0.3) | 2 (0.5) | >0.99 |
| Hypotension | |||
| AEs | 18 (4.5) | 37 (8.8) | 0.02 |
| SAEs | 3 (0.8) | 1 (0.2) | 0.36 |
| Hyperkalaemia | |||
| AEs | 5 (1.3) | 22 (5.2) | 0.001 |
| SAEs | 0 (0) | 0 (0) | — |
| Placebo (n = 397) | Aliskiren (n = 422a) | P-value | |
|---|---|---|---|
| Total adverse events (AEs) | 268 (67.5) | 316 (74.9) | 0.02 |
| Total serious adverse events (SAEs) | 92 (23.2) | 107 (25.4) | 0.51 |
| Renal dysfunction | |||
| AEs | 3 (0.8) | 10 (2.4) | 0.09 |
| SAEs | 1 (0.3) | 2 (0.5) | >0.99 |
| Hypotension | |||
| AEs | 18 (4.5) | 37 (8.8) | 0.02 |
| SAEs | 3 (0.8) | 1 (0.2) | 0.36 |
| Hyperkalaemia | |||
| AEs | 5 (1.3) | 22 (5.2) | 0.001 |
| SAEs | 0 (0) | 0 (0) | — |
aResults are based on treated patients. One patient randomized to the aliskiren group did not take study medication.
P-values from Fisher's exact test.
Investigator and patient reported adverse events
| Placebo (n = 397) | Aliskiren (n = 422a) | P-value | |
|---|---|---|---|
| Total adverse events (AEs) | 268 (67.5) | 316 (74.9) | 0.02 |
| Total serious adverse events (SAEs) | 92 (23.2) | 107 (25.4) | 0.51 |
| Renal dysfunction | |||
| AEs | 3 (0.8) | 10 (2.4) | 0.09 |
| SAEs | 1 (0.3) | 2 (0.5) | >0.99 |
| Hypotension | |||
| AEs | 18 (4.5) | 37 (8.8) | 0.02 |
| SAEs | 3 (0.8) | 1 (0.2) | 0.36 |
| Hyperkalaemia | |||
| AEs | 5 (1.3) | 22 (5.2) | 0.001 |
| SAEs | 0 (0) | 0 (0) | — |
| Placebo (n = 397) | Aliskiren (n = 422a) | P-value | |
|---|---|---|---|
| Total adverse events (AEs) | 268 (67.5) | 316 (74.9) | 0.02 |
| Total serious adverse events (SAEs) | 92 (23.2) | 107 (25.4) | 0.51 |
| Renal dysfunction | |||
| AEs | 3 (0.8) | 10 (2.4) | 0.09 |
| SAEs | 1 (0.3) | 2 (0.5) | >0.99 |
| Hypotension | |||
| AEs | 18 (4.5) | 37 (8.8) | 0.02 |
| SAEs | 3 (0.8) | 1 (0.2) | 0.36 |
| Hyperkalaemia | |||
| AEs | 5 (1.3) | 22 (5.2) | 0.001 |
| SAEs | 0 (0) | 0 (0) | — |
aResults are based on treated patients. One patient randomized to the aliskiren group did not take study medication.
P-values from Fisher's exact test.
Biochemical abnormalities
| Biochemical abnormalities | Placebo, n = 397 (%) | Aliskiren, n = 422 (%) | P-value |
|---|---|---|---|
| Blood urea nitrogen | |||
| >14.3 mmol/L (40 mg/dL) | 18 (4.6) | 52 (12.4) | <0.001 |
| Creatininea | |||
| >176 and <265 μmol/L (>2 and <3 mg/dL) | 4 (1.0) | 13 (3.1) | 0.041a |
| ≥265 μmol/L (≥3 mg/dL) | 1 (0.3) | 2 (0.5) | >0.999a |
| Potassiumb | |||
| <3.5 mmol/L | 11 (2.8) | 10 (2.4) | 0.826b |
| >5.5 and <6.0 mmol/L | 16 (4.0) | 32 (7.6) | 0.002b |
| ≥6.0 mmol/L | 10 (2.5) | 23 (5.5) | 0.034b |
| Biochemical abnormalities | Placebo, n = 397 (%) | Aliskiren, n = 422 (%) | P-value |
|---|---|---|---|
| Blood urea nitrogen | |||
| >14.3 mmol/L (40 mg/dL) | 18 (4.6) | 52 (12.4) | <0.001 |
| Creatininea | |||
| >176 and <265 μmol/L (>2 and <3 mg/dL) | 4 (1.0) | 13 (3.1) | 0.041a |
| ≥265 μmol/L (≥3 mg/dL) | 1 (0.3) | 2 (0.5) | >0.999a |
| Potassiumb | |||
| <3.5 mmol/L | 11 (2.8) | 10 (2.4) | 0.826b |
| >5.5 and <6.0 mmol/L | 16 (4.0) | 32 (7.6) | 0.002b |
| ≥6.0 mmol/L | 10 (2.5) | 23 (5.5) | 0.034b |
P-values from Fisher's exact test.
aCreatinine tests for ≤176 vs. >176 and ≤265 vs. >265 μmol/L.
bPotassium tests for <3.5 vs. ≥3.5, ≤5.5 vs. >5.5, and <6 vs. ≥6 mmol/L.
Biochemical abnormalities
| Biochemical abnormalities | Placebo, n = 397 (%) | Aliskiren, n = 422 (%) | P-value |
|---|---|---|---|
| Blood urea nitrogen | |||
| >14.3 mmol/L (40 mg/dL) | 18 (4.6) | 52 (12.4) | <0.001 |
| Creatininea | |||
| >176 and <265 μmol/L (>2 and <3 mg/dL) | 4 (1.0) | 13 (3.1) | 0.041a |
| ≥265 μmol/L (≥3 mg/dL) | 1 (0.3) | 2 (0.5) | >0.999a |
| Potassiumb | |||
| <3.5 mmol/L | 11 (2.8) | 10 (2.4) | 0.826b |
| >5.5 and <6.0 mmol/L | 16 (4.0) | 32 (7.6) | 0.002b |
| ≥6.0 mmol/L | 10 (2.5) | 23 (5.5) | 0.034b |
| Biochemical abnormalities | Placebo, n = 397 (%) | Aliskiren, n = 422 (%) | P-value |
|---|---|---|---|
| Blood urea nitrogen | |||
| >14.3 mmol/L (40 mg/dL) | 18 (4.6) | 52 (12.4) | <0.001 |
| Creatininea | |||
| >176 and <265 μmol/L (>2 and <3 mg/dL) | 4 (1.0) | 13 (3.1) | 0.041a |
| ≥265 μmol/L (≥3 mg/dL) | 1 (0.3) | 2 (0.5) | >0.999a |
| Potassiumb | |||
| <3.5 mmol/L | 11 (2.8) | 10 (2.4) | 0.826b |
| >5.5 and <6.0 mmol/L | 16 (4.0) | 32 (7.6) | 0.002b |
| ≥6.0 mmol/L | 10 (2.5) | 23 (5.5) | 0.034b |
P-values from Fisher's exact test.
aCreatinine tests for ≤176 vs. >176 and ≤265 vs. >265 μmol/L.
bPotassium tests for <3.5 vs. ≥3.5, ≤5.5 vs. >5.5, and <6 vs. ≥6 mmol/L.
Discussion
In patients with LV systolic dysfunction after recent acute MI, the addition of the direct renin inhibitor aliskiren to a standard optimal medical regimen, including an ACE-I or an ARB, a beta-blocker and in some cases an aldosterone antagonist, did not improve ventricular remodelling compared with placebo.
It is unlikely that this lack of effect on remodelling can be explained by the lack of incremental RAAS blockade as the dose of aliskiren we used (and a lower dose) is known to result in further inhibition of the RAAS when added to an ACE-I or ARB, with further lowering of blood pressure in patients with hypertension,10 and further reduction in albuminuria in patients with diabetes, chronic kidney disease and albuminuria.12 In patients with heart failure, addition of aliskiren to the standard therapy, including an ACE-I or an ARB and beta-blocker decreased plasma BNP concentration.11 The increased rate of hypotension and renal dysfunction with dual therapy in the present study is also indicative of incremental RAAS blockade.
Nor is it likely that the specific approach we used to incrementally block the RAAS was inferior to adding an ARB. Aliskiren is similarly efficacious to other inhibitors of the RAAS in lowering blood pressure and urinary albumin excretion in direct comparisons.9 By competitively inhibiting renin at the most proximal step in the RAAS cascade, aliskiren has a number of potential advantages over other RAAS inhibitors. It attenuates the reflex rise in plasma renin activity and angiotensin I and/or angiotensin II that occurs with use of an ACE-I or ARB and thus may prevent reactivation of the RAAS. Plasma renin activity itself may have direct detrimental biological effects. In support of this hypothesis is the finding that plasma renin activity is a predictor of adverse outcomes in patients with heart failure,16 and following myocardial infarction,17 even in the setting of ACE-I.18,19
The more likely, and simpler, explanation for our findings is that, in contrast to chronic heart failure,20 in the post-MI setting a single inhibitor of the renin–angiotensin system is sufficient to attenuate late remodelling in otherwise well-treated patients, including use of a beta-blocker in nearly every case. Indeed, we cannot exclude the possibility that beta-blockers, known inhibitors of renin release,21 contributed to these findings. Nevertheless, these findings contrast with those of post-MI studies in similar populations with aldosterone receptor antagonists,14,22 suggesting that adding an aldosterone receptor antagonist to an inhibitor of the renin–angiotensin system may be fundamentally different from adding an upstream inhibitor to an ACE-I or ARB.
We found a borderline statistical interaction between baseline diabetes status and treatment effect, such that patients with diabetes-derived greater benefit from aliskiren with respect to the primary outcome. These findings are similar to those reported in the Aliskiren in Left Ventricular Hypertrophy (ALLAY) Trial in which diabetic patients treated with a combination of aliskiren and losartan demonstrated greater left ventricular mass regression than patients treated with losartan alone.23 Nevertheless, these post hoc findings should be considered hypothesis generating.
Some limitations of this study should be noted. Because patients were stabilized on optimal medical therapy prior to initiation of the study drug, which occurred an average of 43 days post-MI, this study had no opportunity to affect early post-MI remodelling, during which time the rate of ventricular remodelling is greatest, although, as found in VALIANT, it is unlikely that more intense RAAS blockade could be tolerated in the acute phase in ACE-I naïve patients. Furthermore, an echocardiographic substudy in VALIANT showed no remodelling benefit of dual therapy started in the early post-MI period24 and a recent study showed that aliskiren use in acute coronary syndrome patients did not reduce levels of brain natriuretic peptide.25 The duration of our study was relatively short, and it is possible that longer administration of aliskiren might be necessary to demonstrate a benefit, although the present findings suggest a measureable between-treatment difference would not be apparent for a very long time.
Consequently, we believe that our trial fulfilled its objective in helping determine whether a large-scale, mortality-morbidity, trial using aliskiren in addition to an ACE-I or ARB should be undertaken in high-risk post-MI patients, and that these findings do not support such a study. On the other hand, our findings about the potential value of dual RAAS blockade with aliskiren should not be extrapolated to other populations, including diabetic nephropathy and heart failure, where the effect on surrogate outcomes has already been shown, and the effect on clinical outcomes will be determined in ongoing clinical trials.26,27
In summary, in a clinical trial comparing aliskiren to placebo in high-risk post-MI patients on optimal medical therapy including an inhibitor of the renin–angiotensin system, we did not observe a benefit with respect to attenuation of ventricular remodelling. Patients receiving aliskiren had overall more adverse events, including hypotension, renal dysfunction, and hyperkalaemia. These findings do not suggest that dual RAAS blockade with aliskiren would provide additional benefit in these high-risk post-MI patients.
Supplementary material
Funding
Funded by Novartis.
Conflict of interest: S.D.S, M.A.P., L.K., J.L.R., A.P.M., J.J.V.M., and A.D. have received research support from Novartis. S.D.S, M.A.P., L.K., J.L.R., A.P.M., J.J.V.M. have consulted for Novartis. R.Y.K. and A.H. are employees of Novartis.


{kind=link}
{kind=link}
{kind=link}