





Abstract
Statins are essential for the reduction of risk of coronary heart disease (CHD) in familial hypercholesterolemia (FH). One of many genes influenced by statin treatment is the ATP-binding cassette transporter A1 (ABCA1) gene, which plays an important role in metabolism of high-density lipoprotein (HDL). The present aim was to test if the ABCA1 C69T polymorphism influences CHD risk and response to statin treatment.
In a large cohort of 1686 FH patients without a history of CHD before 1 January 1990, we analysed statin-ABCA1 C69T polymorphism interaction by comparing treated and untreated patients. We used a Cox proportional hazard model adjusted for sex, birth year, and smoking. In analyses restricted to untreated patients, the TT genotype was associated with 1.7 times higher CHD risk than the CC genotype (hazard ratio (HR) =1.65, 95% confidence interval (95% CI): 1.08–2.53; P = 0.02). Conversely, in statin-treated FH patients, CHD risk in TT individuals was not increased (HR: 0.65, 95% CI: 0.35–1.24; P = 0.2). Formal testing confirmed this interaction (P = 0.03). HDL-cholesterol levels were significantly more raised in statin-treated patients with the TT than with the CC genotype (two-way ANOVA, P = 0.045).
In untreated FH patients, the TT genotype of the ABCA1 C69T polymorphism was associated with increased CHD risk. However, in statin-treated patients, CHD risk was no longer significantly different between genotypes, at least partially explained by a higher rise in HDL-cholesterol levels during statin treatment in TT individuals.
Introduction
Familial hypercholesterolemia (FH) is a hereditary disorder of lipoprotein metabolism associated with a severely increased risk of coronary heart disease (CHD).1 In heterozygous FH, statin treatment reduces CHD risk substantially.2–4 Statins effectively inhibit endogenous cholesterol synthesis, which results in upregulation of low-density lipoprotein (LDL) receptor expression on the cell surface of hepatocytes. Next to this, statins have a number of additional beneficial effects. These so-called pleitropic effects, such as raising high-density lipoprotein cholesterol (HDL-C) levels, increasing cholesterol efflux from cholesterol loaded macrophages, and inhibiting inflammation may explain that CHD risk reduction by statins is greater than expected from LDL-cholesterol (LDL-C) lowering alone.5–7
The genetic background of an individual may influence the response to statin treatment of lipid levels as well as CHD risk. However, not many convincing genetic modifiers of statin response have been identified so far.8–10 Since most FH patients receive statin treatment upon diagnosis, this situation provides a good opportunity to study gene-treatment interactions in these patients.
An interesting candidate gene is the ATP-binding cassette transporter A1 (ABCA1) gene. Hepatic ABCA1 mediates HDL synthesis and ABCA1 is an efficient exporter of cholesterol from macrophages and other cells to HDL in plasma. It is therefore a major factor in CHD risk protection.11–16 A number of polymorphisms in this gene associate with HDL-C levels and CHD risk.17–24
Statins inhibit HMG CoA reductase, the rate-limiting enzyme in the biosynthesis of cholesterol. By inhibiting this early step in the mevalonate pathway, production of isoprenoids and oxysterols is inhibited as well, which are small molecules that influence ABCA1 levels via signaling pathways involving nuclear receptors such as liver-X receptor (LXR) and persoxisome proliferator-activated receptor-γ (PPAR-γ).25–28 Therefore, statin treatment might influence effects of genetic variation in ABCA1.12 A promising polymorphism with regard to response to statin treatment is C69T, since the T allele of this polymorphism was associated with increased CHD risk in the placebo arm of a pravastatin trial.17 As earlier shown for a polymorphism in the gene encoding kinesin-like protein 6 (KIF6), an association restricted to placebo-treated individuals may indicate an interaction between genotype and treatment response.29 Therefore, we hypothesized that ABCA1 C69T TT individuals have an increased risk of CHD, but statin therapy can reduce CHD risk more effectively in TT individuals than in wild-type individuals. The best setting to study gene–statin interaction is a placebo-controlled clinical trial. For ethical reasons these are, however, not allowed in FH patients, which prompted us earlier to simulate such a trial in a large cohort of FH patients to determine the efficacy of statins in primary prevention.4 In the current study, we used the variation in delay of starting statin treatment and the fact that a considerable number of patients refrained from therapy to study gene–statin interaction.
In this large cohort study, we investigated the interaction between the ABCA1 C69T polymorphism and statin treatment on CHD risk in three ways: first, we analysed the association after stratification by statin treatment; secondly, we compared the efficacy of statin treatment between the different genotypes with a Cox regression model in which statin treatment was a time-dependent variable; and finally, we calculated the relative excess risk due to interaction (RERI) as a formal test of interaction.30
Methods
Study population
During 1989–2002, we recruited a cohort of 2400 FH patients from 27 lipid clinics in The Netherlands as described in detail previously.31 In short, 4000 patients were randomly selected from the Dutch DNA database containing over 9300 samples from hypercholesterolaemic patients from more than 60 lipid clinics. From these 4000 patients, 2400 fulfilled the diagnostic criteria for FH. Medical records were reviewed to establish extensive phenotypic data.32 All DNA samples were screened for the presence of LDL-receptor mutations. All patients gave informed consent, and the ethics institutional review board of each participating hospital approved the protocol.
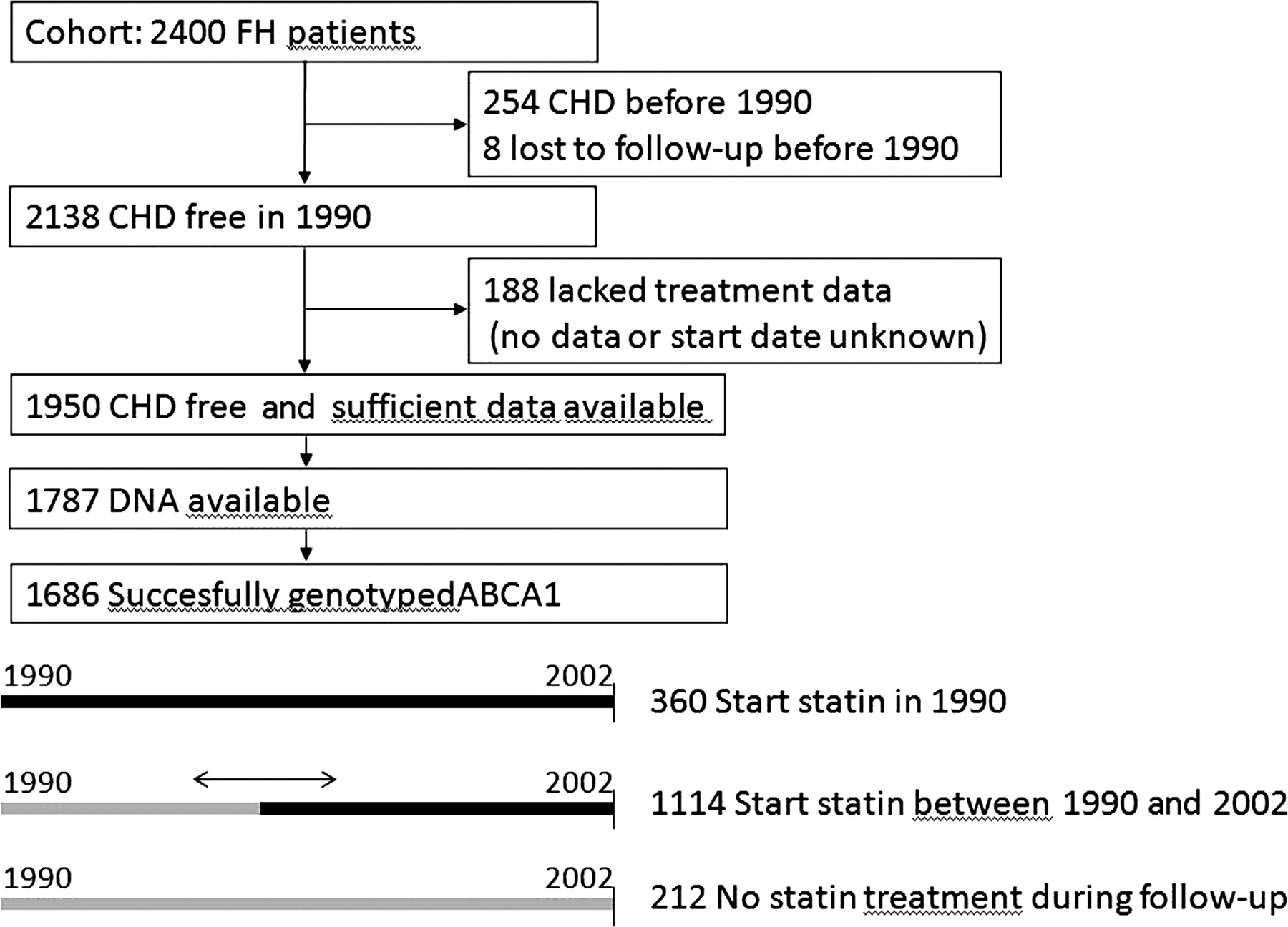
In the present study, to simulate a clinical trial, we chose 1 January 1990, as a starting point, just after the first statin (simvastatin) became available in The Netherlands. We excluded 254 patients, who had prevalent CHD before 1990 to mimic a primary prevention trial (Figure 1).4 Another 188 patients were excluded from the analyses because the type of treatment or the date of starting was unknown. Sufficient data on statin use were available of 1950 event-free FH patients. Of these, DNA was available for 1787 patients. CHD was defined as myocardial infarction, percutaneous coronary intervention, coronary artery bypass grafting, or angina according to earlier described criteria.31 To determine untreated lipid levels, patients were asked to quit cholesterol lowering medication for 6 weeks.
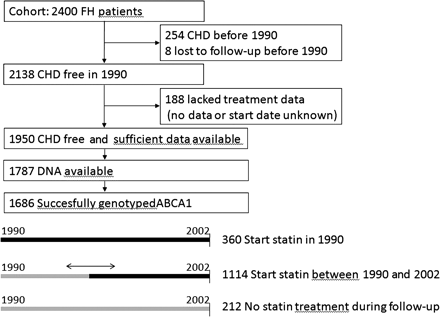
Study design: flow diagram to show included and excluded patients, and the different possibilities for starting point of statin treatment.
Genetic analyses
Genotyping of the C69T polymorphism (rs1800977) was performed by Roche Molecular Systems, CA, USA.22
Statistical analyses
General characteristics were compared using analysis of variance (ANOVA) for continuous variables and χ2 test for categorical variables. Plasma total cholesterol, HDL-C, LDL-C, and triglyceride levels were not normally distributed and were log transformed. Differences in treatment response of lipid levels were analysed with two-way ANOVA analysis. Deviation from Hardy Weinberg Equilibrium was tested with a χ2 test using 1 degree of freedom.
Cox-proportional hazard model was used to model the association between the ABCA1 polymorphism and CHD. Event-free survival time was defined as the period from 1 January 1990 to the date of first CHD or death or censoring at the end of follow-up. Homozygosity for the common allele was assigned as reference.
To compare the influence of statin treatment on association between the polymorphism and CHD, we first calculated HR separately in two strata defined by statin treatment. We considered persons who started statin treatment less than 1 month before an event occurred as untreated.
Next, we stratified by genotype and analysed the influence of statin treatment using statin treatment as a time-dependent variable since most patients experienced a variable period without statin treatment. This variable was equal to 0 for the time no statin treatment was used and 1 for the time from start of statin treatment to the date of first CHD or censoring. Therefore, treated and untreated person-years rather than persons were compared. All analyses were adjusted for year of birth, sex, and smoking.
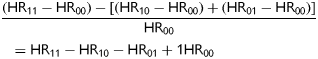
All analyses were performed using SPSS statistical software (Version 14.0, SPSS Inc., Chicago, Ill), except the additional test for interaction, which was performed using SAS software (Version 9.1.3 SAS institute Inc., Cary, NC).
Results
We successfully genotyped 1686 patients for the ABCA1 C69T polymorphism. ABCA1 C69T was in Hardy Weinberg equilibrium (P = 0.86). The genotype frequencies were CC 46.8, CT 43.4, and TT 12.0%. This is comparable to the genotype frequencies found in the general population: CC 46.9, CT 40.7, and TT 12.4% (HapMap Phase 3). The general characteristics including lipid levels were not significantly different between genotypes (Table 1) except diabetes mellitus, which was most common in CC individuals (P = 0.03). In a total of 926 patients, an LDL receptor mutation was identified, the remaining 760 had FH based on clinical criteria.31 The LDL receptor mutations were equally distributed amongst the three ABCA1 C69T genotypes and the mutation frequencies were similar to those of our nation-wide FH screening programme.33
General characteristics and treatment parameters per genotype
| ABCA1 C69T genotype | ||||
|---|---|---|---|---|
| CC | CT | TT | P-value | |
| Number | 752 | 732 | 202 | |
| Male (%) | 352 (46.8) | 351 (48.0) | 88 (43.6) | 0.54 |
| Age, January 1990 | 39.6 ± 12.8 | 38.9 ± 12.9 | 38.1 ± 12.4 | 0.26 |
| Smoking ever (%) | 511 (75.5) | 484 (73.0) | 127 (70.6) | 0.34 |
| Hypertension ever (%) | 70 (9.4) | 58 (8) | 16 (8) | 0.60 |
| Diabetes mellitus ever (%) | 46 (6.1) | 25 (3.4) | 7 (3.5) | 0.03 |
| Cardiac event (%) | 167 (22.2) | 151 (20.6) | 42 (20.8) | 0.74 |
| BMI (kg/m2) | 25.0 ± 3.4 | 25.1 ± 3.7 | 24.6 ± 3 | 0.30 |
| Statin, January 1990 (%) | 164 (28.7) | 160 (28.6) | 31 (20.5) | 0.11 |
| Simvastatin (%) | 436 (76.4) | 457 (81.6) | 118 (78.1) | 0.68 |
| Dosage (mg) | 31 ± 20 | 32 ± 19 | 34 ± 20 | 0.40 |
| Age of starting statin | 43.2 ± 125 | 42.5 ± 12.8 | 42.6 ± 12.1 | 0.60 |
| Lipid levels | ||||
| Without treatment | ||||
| Total cholesterol (mmol/L) | 9.21 ± 1.80 | 9.17 ± 1.82 | 9.09 ± 1.86 | 0.69 |
| LDL-C (mmol/L) | 7.04 ± 1.72 | 7.02 ± 1.78 | 6.92 ± 1.78 | 0.72 |
| HDL-C (mmol/L) | 1.17 ± 0.33 | 1.18 ± 0.33 | 1.18 ± 0.34 | 0.88 |
| Triglycerides (mmol/L) | 1.56 ± 0.79 | 1.56 ± 0.79 | 1.60 ± 0.79 | 0.78 |
| During statin treatment | ||||
| Total cholesterol (mmol/L) | 5.93 ± 1.24 | 5.97 ± 1.25 | 5.89 ± 1.25 | 0.60 |
| LDL-C (mmol/L) | 3.93 ± 1.17 | 4.0 ± 1.17 | 3.82 ± 1.34 | 0.14 |
| HDL-C (mmol/L) | 1.27 ± 0.35 | 1.29 ± 0.35 | 1.34 ± 0.38 | 0.41 |
| Triglycerids (mmol/L) | 1.23 ± 0.60 | 1.1 ± 0.55 | 1.19 ± 0.66 | 0.30 |
| ABCA1 C69T genotype | ||||
|---|---|---|---|---|
| CC | CT | TT | P-value | |
| Number | 752 | 732 | 202 | |
| Male (%) | 352 (46.8) | 351 (48.0) | 88 (43.6) | 0.54 |
| Age, January 1990 | 39.6 ± 12.8 | 38.9 ± 12.9 | 38.1 ± 12.4 | 0.26 |
| Smoking ever (%) | 511 (75.5) | 484 (73.0) | 127 (70.6) | 0.34 |
| Hypertension ever (%) | 70 (9.4) | 58 (8) | 16 (8) | 0.60 |
| Diabetes mellitus ever (%) | 46 (6.1) | 25 (3.4) | 7 (3.5) | 0.03 |
| Cardiac event (%) | 167 (22.2) | 151 (20.6) | 42 (20.8) | 0.74 |
| BMI (kg/m2) | 25.0 ± 3.4 | 25.1 ± 3.7 | 24.6 ± 3 | 0.30 |
| Statin, January 1990 (%) | 164 (28.7) | 160 (28.6) | 31 (20.5) | 0.11 |
| Simvastatin (%) | 436 (76.4) | 457 (81.6) | 118 (78.1) | 0.68 |
| Dosage (mg) | 31 ± 20 | 32 ± 19 | 34 ± 20 | 0.40 |
| Age of starting statin | 43.2 ± 125 | 42.5 ± 12.8 | 42.6 ± 12.1 | 0.60 |
| Lipid levels | ||||
| Without treatment | ||||
| Total cholesterol (mmol/L) | 9.21 ± 1.80 | 9.17 ± 1.82 | 9.09 ± 1.86 | 0.69 |
| LDL-C (mmol/L) | 7.04 ± 1.72 | 7.02 ± 1.78 | 6.92 ± 1.78 | 0.72 |
| HDL-C (mmol/L) | 1.17 ± 0.33 | 1.18 ± 0.33 | 1.18 ± 0.34 | 0.88 |
| Triglycerides (mmol/L) | 1.56 ± 0.79 | 1.56 ± 0.79 | 1.60 ± 0.79 | 0.78 |
| During statin treatment | ||||
| Total cholesterol (mmol/L) | 5.93 ± 1.24 | 5.97 ± 1.25 | 5.89 ± 1.25 | 0.60 |
| LDL-C (mmol/L) | 3.93 ± 1.17 | 4.0 ± 1.17 | 3.82 ± 1.34 | 0.14 |
| HDL-C (mmol/L) | 1.27 ± 0.35 | 1.29 ± 0.35 | 1.34 ± 0.38 | 0.41 |
| Triglycerids (mmol/L) | 1.23 ± 0.60 | 1.1 ± 0.55 | 1.19 ± 0.66 | 0.30 |
BMI, body mass index; values are mean ± standard deviation.
General characteristics and treatment parameters per genotype
| ABCA1 C69T genotype | ||||
|---|---|---|---|---|
| CC | CT | TT | P-value | |
| Number | 752 | 732 | 202 | |
| Male (%) | 352 (46.8) | 351 (48.0) | 88 (43.6) | 0.54 |
| Age, January 1990 | 39.6 ± 12.8 | 38.9 ± 12.9 | 38.1 ± 12.4 | 0.26 |
| Smoking ever (%) | 511 (75.5) | 484 (73.0) | 127 (70.6) | 0.34 |
| Hypertension ever (%) | 70 (9.4) | 58 (8) | 16 (8) | 0.60 |
| Diabetes mellitus ever (%) | 46 (6.1) | 25 (3.4) | 7 (3.5) | 0.03 |
| Cardiac event (%) | 167 (22.2) | 151 (20.6) | 42 (20.8) | 0.74 |
| BMI (kg/m2) | 25.0 ± 3.4 | 25.1 ± 3.7 | 24.6 ± 3 | 0.30 |
| Statin, January 1990 (%) | 164 (28.7) | 160 (28.6) | 31 (20.5) | 0.11 |
| Simvastatin (%) | 436 (76.4) | 457 (81.6) | 118 (78.1) | 0.68 |
| Dosage (mg) | 31 ± 20 | 32 ± 19 | 34 ± 20 | 0.40 |
| Age of starting statin | 43.2 ± 125 | 42.5 ± 12.8 | 42.6 ± 12.1 | 0.60 |
| Lipid levels | ||||
| Without treatment | ||||
| Total cholesterol (mmol/L) | 9.21 ± 1.80 | 9.17 ± 1.82 | 9.09 ± 1.86 | 0.69 |
| LDL-C (mmol/L) | 7.04 ± 1.72 | 7.02 ± 1.78 | 6.92 ± 1.78 | 0.72 |
| HDL-C (mmol/L) | 1.17 ± 0.33 | 1.18 ± 0.33 | 1.18 ± 0.34 | 0.88 |
| Triglycerides (mmol/L) | 1.56 ± 0.79 | 1.56 ± 0.79 | 1.60 ± 0.79 | 0.78 |
| During statin treatment | ||||
| Total cholesterol (mmol/L) | 5.93 ± 1.24 | 5.97 ± 1.25 | 5.89 ± 1.25 | 0.60 |
| LDL-C (mmol/L) | 3.93 ± 1.17 | 4.0 ± 1.17 | 3.82 ± 1.34 | 0.14 |
| HDL-C (mmol/L) | 1.27 ± 0.35 | 1.29 ± 0.35 | 1.34 ± 0.38 | 0.41 |
| Triglycerids (mmol/L) | 1.23 ± 0.60 | 1.1 ± 0.55 | 1.19 ± 0.66 | 0.30 |
| ABCA1 C69T genotype | ||||
|---|---|---|---|---|
| CC | CT | TT | P-value | |
| Number | 752 | 732 | 202 | |
| Male (%) | 352 (46.8) | 351 (48.0) | 88 (43.6) | 0.54 |
| Age, January 1990 | 39.6 ± 12.8 | 38.9 ± 12.9 | 38.1 ± 12.4 | 0.26 |
| Smoking ever (%) | 511 (75.5) | 484 (73.0) | 127 (70.6) | 0.34 |
| Hypertension ever (%) | 70 (9.4) | 58 (8) | 16 (8) | 0.60 |
| Diabetes mellitus ever (%) | 46 (6.1) | 25 (3.4) | 7 (3.5) | 0.03 |
| Cardiac event (%) | 167 (22.2) | 151 (20.6) | 42 (20.8) | 0.74 |
| BMI (kg/m2) | 25.0 ± 3.4 | 25.1 ± 3.7 | 24.6 ± 3 | 0.30 |
| Statin, January 1990 (%) | 164 (28.7) | 160 (28.6) | 31 (20.5) | 0.11 |
| Simvastatin (%) | 436 (76.4) | 457 (81.6) | 118 (78.1) | 0.68 |
| Dosage (mg) | 31 ± 20 | 32 ± 19 | 34 ± 20 | 0.40 |
| Age of starting statin | 43.2 ± 125 | 42.5 ± 12.8 | 42.6 ± 12.1 | 0.60 |
| Lipid levels | ||||
| Without treatment | ||||
| Total cholesterol (mmol/L) | 9.21 ± 1.80 | 9.17 ± 1.82 | 9.09 ± 1.86 | 0.69 |
| LDL-C (mmol/L) | 7.04 ± 1.72 | 7.02 ± 1.78 | 6.92 ± 1.78 | 0.72 |
| HDL-C (mmol/L) | 1.17 ± 0.33 | 1.18 ± 0.33 | 1.18 ± 0.34 | 0.88 |
| Triglycerides (mmol/L) | 1.56 ± 0.79 | 1.56 ± 0.79 | 1.60 ± 0.79 | 0.78 |
| During statin treatment | ||||
| Total cholesterol (mmol/L) | 5.93 ± 1.24 | 5.97 ± 1.25 | 5.89 ± 1.25 | 0.60 |
| LDL-C (mmol/L) | 3.93 ± 1.17 | 4.0 ± 1.17 | 3.82 ± 1.34 | 0.14 |
| HDL-C (mmol/L) | 1.27 ± 0.35 | 1.29 ± 0.35 | 1.34 ± 0.38 | 0.41 |
| Triglycerids (mmol/L) | 1.23 ± 0.60 | 1.1 ± 0.55 | 1.19 ± 0.66 | 0.30 |
BMI, body mass index; values are mean ± standard deviation.
In January 1990, 360 of these patients were treated with a statin, whereas during follow-up 1113 patients were prescribed statin treatment as well (Figure 1). Of the statin-treated patients, 79% was treated with simvastatin (mean dosage of 33 mg), 13% with atorvastatin (mean dosage 48 mg), 6% pravastatin (mean dosage 30 mg), and 2% with other statins. The age of starting statin treatment was not significantly different between genotypes (Table 1). Twenty-one patients discontinued statin treatment for unknown reasons.
During a total follow-up of 14 085 person-years, 360 CHD events occurred. Of these, 139 patients had been receiving statins before their events during a median period of 2.3 years; the other 221 events occurred in patients who had not yet been treated with statins. Eleven patients had DM prior to their events. As we showed earlier, statin treatment reduced CHD risk by approximately 80% in a Cox regression model using statin treatment as a time-dependent variable.4 In the complete population, including treated as well as untreated patients, the ABCA1 variant was not associated with CHD risk (TT vs. CC HR: 1.01, 95% confidence interval (95% CI): 0.72–1.41; P = 0.97).
CHD risk according to ABCA1 C69T genotypes in statin-treated and untreated patients is shown in Table 2. Untreated TT individuals had an increased CHD risk compared with untreated CC individuals (HR: 1.65, 95% CI: 1.08–2.53; P = 0.02) but this was not the case in treated patients (HR: 0.65, 95% CI: 0.35–1.24; P = 0.2). CT individuals had a CHD risk in between CC and TT individuals; the P for trend was 0.042 in untreated individuals but not significant in treated individuals (Table 2).
Association of ATP-binding cassette transporter A1 C69T genotype and coronary heart disease risk without or during statin treatment
| ABCA-1 C69T | ||||
|---|---|---|---|---|
| Genotype | n | HR (95% CI) | P-value | |
| Not using statins | CC | 181 | (ref) | |
| CT | 172 | 1.11 (0.83–1.50) | 0.48 | |
| TT | 51 | 1.65 (1.08–2.53) | 0.02 | |
| During statin treatment | CC | 571 | (ref) | |
| CT | 560 | 0.92 (0.64–1.33) | 0.66 | |
| TT | 151 | 0.65 (0.34–1.24) | 0.19 | |
| ABCA-1 C69T | ||||
|---|---|---|---|---|
| Genotype | n | HR (95% CI) | P-value | |
| Not using statins | CC | 181 | (ref) | |
| CT | 172 | 1.11 (0.83–1.50) | 0.48 | |
| TT | 51 | 1.65 (1.08–2.53) | 0.02 | |
| During statin treatment | CC | 571 | (ref) | |
| CT | 560 | 0.92 (0.64–1.33) | 0.66 | |
| TT | 151 | 0.65 (0.34–1.24) | 0.19 | |
HR, hazard ratio; 95% CI, 95% confidence interval; adjusted for sex, smoking, year of birth; CC as reference.
Association of ATP-binding cassette transporter A1 C69T genotype and coronary heart disease risk without or during statin treatment
| ABCA-1 C69T | ||||
|---|---|---|---|---|
| Genotype | n | HR (95% CI) | P-value | |
| Not using statins | CC | 181 | (ref) | |
| CT | 172 | 1.11 (0.83–1.50) | 0.48 | |
| TT | 51 | 1.65 (1.08–2.53) | 0.02 | |
| During statin treatment | CC | 571 | (ref) | |
| CT | 560 | 0.92 (0.64–1.33) | 0.66 | |
| TT | 151 | 0.65 (0.34–1.24) | 0.19 | |
| ABCA-1 C69T | ||||
|---|---|---|---|---|
| Genotype | n | HR (95% CI) | P-value | |
| Not using statins | CC | 181 | (ref) | |
| CT | 172 | 1.11 (0.83–1.50) | 0.48 | |
| TT | 51 | 1.65 (1.08–2.53) | 0.02 | |
| During statin treatment | CC | 571 | (ref) | |
| CT | 560 | 0.92 (0.64–1.33) | 0.66 | |
| TT | 151 | 0.65 (0.34–1.24) | 0.19 | |
HR, hazard ratio; 95% CI, 95% confidence interval; adjusted for sex, smoking, year of birth; CC as reference.
We further analysed the gene–drug interaction comparing both treated and untreated carriers of the different genotypes using untreated CC individuals as reference (Table 3). Relative to CC individuals without treatment, statin treatment reduced CHD risk in CC individuals by 85% (HR: 0.15, 95% CI: 0.11–0.21; P < 0.001) and in TT individuals by 90% (HR: 0.097, 95% CI: 0.05–0.181; P < 0.001). This indicates a larger CHD risk reduction by statin treatment of TT individuals as 75% (HR: 0.25) instead of 90% (HR: 0.097) was expected if there had been no interaction between genotype and treatment. We calculated the expected HR by multiplying the HR of CHD risk reduction by statin treatment of CC individuals by CHD risk in untreated TT individuals (0.15 × 1.65 = 0.25).
Interaction between genotype and statin treatment on coronary heart disease risk
| ABCA-1 C69T | ||||
|---|---|---|---|---|
| Statin treatment | Genotype | n | HR (95% CI) | P-value |
| No | CC | 181 | (ref) | |
| Current | 571 | 0.15 (0.11–0.21) | <0.001 | |
| No | CT | 172 | 1.09 (0.81–1.47) | 0.56 |
| Current | 560 | 0.14 (0.10–0.19) | <0.001 | |
| No | TT | 51 | 1.65 (1.08–2.53) | 0.02 |
| Current | 151 | 0.10 (0.05–0.18) | <0.001 | |
| ABCA-1 C69T | ||||
|---|---|---|---|---|
| Statin treatment | Genotype | n | HR (95% CI) | P-value |
| No | CC | 181 | (ref) | |
| Current | 571 | 0.15 (0.11–0.21) | <0.001 | |
| No | CT | 172 | 1.09 (0.81–1.47) | 0.56 |
| Current | 560 | 0.14 (0.10–0.19) | <0.001 | |
| No | TT | 51 | 1.65 (1.08–2.53) | 0.02 |
| Current | 151 | 0.10 (0.05–0.18) | <0.001 | |
Adjusted for sex, smoking, year of birth; CC without treatment as reference.
Interaction between genotype and statin treatment on coronary heart disease risk
| ABCA-1 C69T | ||||
|---|---|---|---|---|
| Statin treatment | Genotype | n | HR (95% CI) | P-value |
| No | CC | 181 | (ref) | |
| Current | 571 | 0.15 (0.11–0.21) | <0.001 | |
| No | CT | 172 | 1.09 (0.81–1.47) | 0.56 |
| Current | 560 | 0.14 (0.10–0.19) | <0.001 | |
| No | TT | 51 | 1.65 (1.08–2.53) | 0.02 |
| Current | 151 | 0.10 (0.05–0.18) | <0.001 | |
| ABCA-1 C69T | ||||
|---|---|---|---|---|
| Statin treatment | Genotype | n | HR (95% CI) | P-value |
| No | CC | 181 | (ref) | |
| Current | 571 | 0.15 (0.11–0.21) | <0.001 | |
| No | CT | 172 | 1.09 (0.81–1.47) | 0.56 |
| Current | 560 | 0.14 (0.10–0.19) | <0.001 | |
| No | TT | 51 | 1.65 (1.08–2.53) | 0.02 |
| Current | 151 | 0.10 (0.05–0.18) | <0.001 | |
Adjusted for sex, smoking, year of birth; CC without treatment as reference.
Comparison of risk reduction by directly comparing untreated and statin-treated person-years using statin use as a time-dependent variable also showed that the use of statins is associated with a higher risk reduction in TT individuals (HR: 0.12, 95% CI: 0.04–0.36; P < 0.001), although 95% confidence intervals overlapped between different genotypes. Risk reduction in CC individuals was 74% (HR: 0.26, 95% CI: 0.18–0.37; P < 0.001). Finally, we confirmed this interaction formally by determination of the RERI (−0.29, 95% CI: −0.03 to −0.55; P = 0.03).
To define whether HDL-C increase in response to statin treatment could explain this difference between untreated and statin-treated patients, we performed a two-way ANOVA according to genotype comparing HDL-C levels with and without statin treatment in all persons who started statin treatment during follow-up (Table 1). TT individuals appeared to experience a larger rise of HDL-C level than CC individuals during statin treatment (13.6% vs. 8.5%; P = 0.045). In Figure 2, HDL-C response to statin treatment and CHD risk in statin-treated patients are shown per genotype. These observations seem to be correlated since CHD risk decreases while HDL-C levels increase. In line with partially explaining the effect on CHD risk, adding HDL-C response to the Cox regression model moved the point estimate towards 1 in treated TT individuals and annihilated the significance (HR: 0.78, 95% CI: 0.38–1.61; P = 0.51). For the model without HDL-C response we refer to Table 2 (HR: 0.65, 95% CI: 0.34–1.24; P = 0.19).
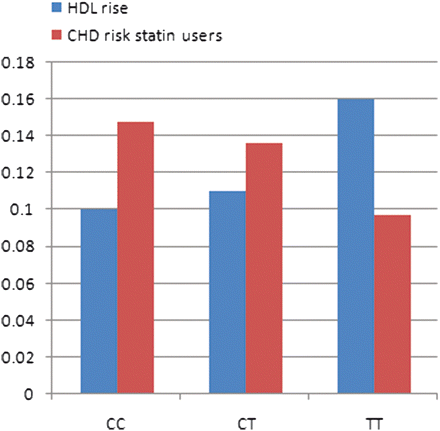
Effect of statin treatment on high-density lipoprotein-cholesterol rise and coronary heart disease risk per genotype. Coronary heart disease risk statin users relative to CC non-users (compare Table 3). Numbers on ordinate either are hazard ratio (coronary heart disease risk) or mmol/L (high-density lipoprotein-cholesterol).
Discussion
In this large cohort study of FH patients, we discovered an interaction between a genetic variant in the ABCA1 gene, statin treatment, and CHD risk. In an untreated situation, the TT genotype of the ABCA1 C69T polymorphism was significantly associated with higher CHD risk in FH patients. Conversely, in statin-treated patients, CHD risk in TT individuals was not increased. HDL-C levels increased to a significantly higher extend in the TT individuals during statin treatment. Taken together, these results suggest that the interaction between the ABCA1 C69T polymorphism and statin treatment changes the impact of the ABCA1 C69T polymorphism on CHD risk, at least partially due to a greater increase of HDL-C in TT individuals. DM was significantly different between genotypes. We do not think this has influenced our results with regard to CHD risk reduction because of the low numbers and the fact that most DM occurred only after a CHD event had taken place. It is however an intriguing finding, reassuring recent studies that found a role for ABCA1 in beta cell function.34 Unfortunately, our study was not sufficiently powered to perform in-depth analyses or interaction studies with treatment.
ABCA1 has an essential role in the regulation of HDL metabolism. The liver is the major source of HDL and hepatocytes require ABCA1 for HDL generation.11 Increased production of HDL may result in an accelerated overall reverse cholesterol transport and thereby be protective against the development of CVD. Moreover, ABCA1 is involved in reverse cholesterol transport itself, controlling the rate-limiting step in cellular cholesterol efflux from macrophages to HDL. Cholesterol efflux from macrophages protects these cells from accumulating cholesterol, thereby delaying the formation of foam cells, one of the early steps in the atherosclerotic process.12–16
The association between the C69T polymorphism, also known as -14T or rs1800977, and CHD risk has been studied before but the results were inconsistent.17–21 A significantly higher number of TT individuals was found amongst Indian CHD patients than amongst Indian controls, but in this study no differences were found in other ethnicities.18 Tregouet et al.19 concluded that there was no association between the polymorphism and CHD. In another small study, Ergen et al.20 observed a non-significant trend towards a higher percentage of TT individuals among MI patients. In contrast, fewer patients with diabetes who had MI were TT carriers compared with those without MI.21 However, one-third of these patients without and 39% of patients with MI were on lipid-lowering drugs. The potential interaction with lipid-lowering medication or more specifically statin treatment was not investigated in these studies. The only study that covered statin treatment was a placebo-controlled randomized trial by Zwarts et al.17: TT individuals had higher CHD risk, but the differences between statin-treated and placebo-treated patients were not specifically reported. In line with our results, CC individuals in the placebo group had lower CHD rates (12.4%) than CT (26.9%) and TT individuals (35.3%). The statin-treated TT group had the fewest events, but the numbers were too small to perform a meaningful statistical analysis.
The association between ABCA1 C69T genotype and HDL-C levels has been studied more extensively but the results are inconsistent as well. In most studies, no relationship was found.17,20,21,25 In both a Turkish and a Chinese population, the TT genotype was associated with higher HDL-C levels in men only, while in a large French cohort this was only observed in men with BMI <25 kg/m2.18,23,24 Remarkably, in another study the TT genotype was associated with lower HDL-C.25 So far, the response of HDL-C levels to statin treatment has not been reported. In our study, baseline HDL-C levels were not affected by the genotypes. Nonetheless, HDL-C was raised to a significantly higher extent by statin treatment in TT individuals than in CC individuals.
Notably, contradicting effects of this polymorphism were also at the molecular level of ABCA1 transcription and translation. The ABCA1 gene uses a number of transcriptional start sites and the predominant transcript differs between cells and tissues.35 As a consequence, ABCA1 transcription rate may differ according to cell type. Also ABCA1 levels might in some but not all tissues may be regulated post-transcriptionally.36 Even more puzzling, the effects of higher ABCA1 expression may differ between cells and tissues and in the presence or absence of hypercholesterolemia.37 Joyce et al.38,39 found opposite effects of ABCA1 expression in macrophages and the liver: while overexpression in macrophages exerts an atheroprotective effect, selective transgenic overexpression of ABCA1 in the liver was atherogenic in LDL receptor knockout mice due to accumulation of cholesterol-enriched apoB-lipoproteins. However, a recent study demonstrated the opposite: Brunham et al.40 showed that deletion of Abca1 in macrophages had no effects on atherosclerosis, but liver-specific deletion of Abca1 in Ldlr−/− mice led to increased atherosclerosis.
The location of the polymorphism in the promoter region between a TATA box and one of the transcriptional start sites suggests that the C69T sequence variant might change transcriptional activity but this effect might also be dependent on factors such as cell type and other factors as described above. In two earlier studies, the TT genotype showed higher transcriptional activity than the CC genotype in different cell lines and in peripheral blood mononuclear cells of hypercholesterolemic patients.23,41 In normocholesterolemic patients no differences were observed.40 The effect of the C69T polymorphism on ABCA1 mRNA and protein levels in the liver remains to be established.
The location of the polymorphism might also explain the sensitivity to molecules influencing transcription such as statins, although this may vary between cell types as well. In peripheral blood mononuclear cells of statin-treated patients, no differences in transcriptional activity in monocytes were observed between CC and TT individuals in contrast to observations in hypercholesterolemic patients.41 Interestingly, statin treatment had different effects on ABCA1 in hepatocytes compared with macrophages: Tamehiro et al.13 found that compactin treatment resulted in upregulation of ABC A1 in the liver, while it downregulated ABCA1 mRNA levels in peripheral cells in rats.
Alternatively, instead of changing gene transcription, the C69T polymorphism may tag a functional polymorphism in the ABCA1 gene. It would be of great interest to study the exact consequences of the C69T polymorphism in more detail, since prediction of treatment response will facilitate personalized medicine. As an example, Celera is now preparing to launch a test for KIF6 to ‘identify those patients at additional risk for an event and those whose incremental risk could be ameliorated by statin therapy’29 (www.celera.com).
The strength of the current study lies in the fact that statin treatment is recommended for all FH patients independently of other risk factors for coronary heart disease, which diminishes the risk of an indication bias. However, treatment was neither randomized nor placebo controlled, which may introduce other kinds of bias. In our earlier study, we showed that the patients who started statin treatment immediately in 1990 had more severe CHD risk profiles: they had more often hypertension, were older and had higher cholesterol levels than the patients who started later. However, this selection bias would have resulted in an underestimation of effect of statin treatment since the most at risk were treated first. In addition, patients might have improved their lifestyle in conjunction with starting statin treatment. Therefore, we analysed the effect of smoking cessation within 6 months after starting statin treatment but this did not change the effect of statin treatment.4 Although these or unrecognized confounding factors may have influenced the determination of efficacy of statin treatment, in the present study we analysed statin–genotype interaction and as a result of ‘Mendelian randomization’ the classical risk factors such as age, smoking, LDL-C, and HDL-C were evenly distributed amongst genotypes. Moreover, the variation in delay of starting statin treatment and the percentage of patients refraining from therapy was similar between genotypes: the genotypes were not related to the decision to start a statin.
In summary, we conclude that the TT genotype of the ABCA1 C69T variant is associated with a higher CHD risk in untreated FH patients. Statin-treated TT individuals, however, have CHD risk similar to the other genotypes. This effect is most likely at least partially caused by a greater increase of HDL-C levels in TT individuals. Our results suggest that statins reduce CHD risk partly through HDL-related mechanisms.
Funding
This work was funded by the Netherlands Heart Foundation (2006B190).
Conflicts of interest: J.J.P.K. has received research funding from, served as a consultant for, and received honoraria for lectures from AstraZeneca, Genzyme, ISIS, Merck, Novartis, Pfizer, Roche, Schering Plough and Sankyo. E.J.G.S. has received not-drug related research funding from Pfizer and Merck.


{kind=link}
{kind=link}
{kind=link}
{kind=link}