





Abstract
Despite improvements in modern cardiovascular therapy, the morbidity and mortality of ischaemic heart disease (IHD) and heart failure (HF) remain significant in Europe and worldwide. Patients with IHD may benefit from therapies that would accelerate natural processes of postnatal collateral vessel formation and/or muscle regeneration. Here, we discuss the use of cells in the context of heart repair, and the most relevant results and current limitations from clinical trials using cell-based therapies to treat IHD and HF. We identify and discuss promising potential new therapeutic strategies that include ex vivo cell-mediated gene therapy, the use of biomaterials and cell-free therapies aimed at increasing the success rates of therapy for IHD and HF. The overall aim of this Position Paper of the ESC Working Group Cellular Biology of the Heart is to provide recommendations on how to improve the therapeutic application of cell-based therapies for cardiac regeneration and repair.
Stem cells in the context of heart repair
Stem cells are defined as cells with the ability (i) to self-renew by dividing to make copies of themselves and (ii) to differentiate to at least one other cell type.1 In the context of cell transplantation and heart repair or regeneration [i.e. replacement or regrowth of heart damaged, respectively, including restoration of the (epicardial) vasculature and neovascularization], the term “stem cells” has been widely used but in retrospect, some of the cells used should not strictly be defined as stem cells. Cells with various molecular and functional properties have been isolated from the heart and termed “cardiac stem cells” (CSCs), “cardiac progenitor cells” (CPCs), or “cardiomyocyte progenitor cells” (CMPCs).2,3 These cells can self-renew in culture, and differentiate into different lineages (endothelial cells and mesenchymal cells), but for example have limited cardiogenic differentiation capacities except under exceptional circumstances. By the addition of compounds that induce demethylation, human CMPCs do form cardiomyocytes.4 Otherwise, the only “stem cells” that form cardiomyocytes using mixtures of growth factors, collectively referred to as “cardiogenic cocktails”, are pluripotent stem cells (PSCs). PSCs can be of embryonic origin (embryonic stem cells, ESCs) or created by introducing reprogramming genes into terminally differentiated cells, to make what are called induced pluripotent stem cells (iPSCs).1 Another term now often regarded as incorrectly used is “endothelial progenitor cells” (EPCs). These cells were originally isolated as populations that grew in culture from peripheral blood samples (reviewed in5). They could form networks that resembled vasculature, but they turned out not to be true mature endothelial cells. Finally, cells that adhere onto tissue culture plastic in serum-containing growth medium and have adipogenic, osteogenic, and chondrogenic differentiation potential in culture were termed mesenchymal stem cells (MSCs).6 However, these cells have not been isolated clonally as single cells and could therefore be heterogeneous cell populations. Moreover, with the exception of MSCs derived from bone marrow, these differentiation effects are not observed in vivo. Thus, despite them all expressing a similar set of surface markers, these cells are now called “bone marrow-derived mesenchymal stromal cells” (BM-MSCs) or adipose tissue-derived mesenchymal stromal cells (AT-MSCs).7 These MSCs have not been shown to spontaneously differentiate into cardiomyocytes. For the purpose of this position paper, we use the terminology as in the (historic) literature for the sake of clarity, but we are aware of the caveats in the terminology itself.
Translation of cell therapy: successful preclinical stories with uncertain clinical efficacy
Ischaemic heart disease (IHD) and heart failure (HF) remain major causes of morbidity and mortality worldwide.8,9 Potentially valid clinical strategies aimed at repairing damaged heart muscle and ischaemic tissue and increasing the heart's regenerative potential, are currently being tested in clinical trials.2,10 Despite originally high expectations fueled by exciting scientific progress in preclinical models, and long-term, randomized clinical trials that showed reassuring safety profiles for intracoronary (IC) delivery of cells,2,11–16 regenerative therapy for cardiovascular disease had been inconsistent and shown modest efficacy thus far.10,17–23 Several limitations of most previous clinical trials of cell-based therapies were raised and should be addressed before we can fully understand their true therapeutic potential (see Table 1).
Limitations of cell-based therapies
| Drawbacks | References |
|---|---|
| 1. Low engraftment of BMCs and blood-derived EPCs | 12,24–26 |
| 2. Poor survival of transplanted cells in ischaemic tissue | 27 |
| 3. Failure of adult stem cells to differentiate efficiently into mature and functional cardiomyocytes | 28 |
| 4. Inadequate recruitment of circulating or resident CSCs | 2,29 |
| 5. Anomalous electromechanical coupling between the transplanted cells or between the transplanted and host cells with consequent arrhythmias | 30 |
| 6. The use of LVEF for assessing the effects of cell therapy, as this is a load-dependent variable and loss of contractility may be compensated by increases in preload and decrease in afterload that determine changes of the Frank-Starling forces | 31 |
| 7. Incorrect target population of not very sick patients with baseline LVEF ∼50%, with generally a favourable outcome | 31 |
| 8. Existence of well-established alternative therapeutic strategies (PCI, FL, ACE-, and β-blockers) that might mask potential of cell therapy effects | 32 |
| 9. The lack of experimental validation (prove of efficacy) of cell preparation within the trial course |
| Drawbacks | References |
|---|---|
| 1. Low engraftment of BMCs and blood-derived EPCs | 12,24–26 |
| 2. Poor survival of transplanted cells in ischaemic tissue | 27 |
| 3. Failure of adult stem cells to differentiate efficiently into mature and functional cardiomyocytes | 28 |
| 4. Inadequate recruitment of circulating or resident CSCs | 2,29 |
| 5. Anomalous electromechanical coupling between the transplanted cells or between the transplanted and host cells with consequent arrhythmias | 30 |
| 6. The use of LVEF for assessing the effects of cell therapy, as this is a load-dependent variable and loss of contractility may be compensated by increases in preload and decrease in afterload that determine changes of the Frank-Starling forces | 31 |
| 7. Incorrect target population of not very sick patients with baseline LVEF ∼50%, with generally a favourable outcome | 31 |
| 8. Existence of well-established alternative therapeutic strategies (PCI, FL, ACE-, and β-blockers) that might mask potential of cell therapy effects | 32 |
| 9. The lack of experimental validation (prove of efficacy) of cell preparation within the trial course |
BMCs, bone marrow cells; EPCs, endothelial progenitor cells; CSCs, cardiac stem cells; LVEF, left ventricle ejection fraction; PCI, percutaneous coronary intervention; FL, fibrinolysis; ACE-, angiotensin-converting enzyme inhibition.
Limitations of cell-based therapies
| Drawbacks | References |
|---|---|
| 1. Low engraftment of BMCs and blood-derived EPCs | 12,24–26 |
| 2. Poor survival of transplanted cells in ischaemic tissue | 27 |
| 3. Failure of adult stem cells to differentiate efficiently into mature and functional cardiomyocytes | 28 |
| 4. Inadequate recruitment of circulating or resident CSCs | 2,29 |
| 5. Anomalous electromechanical coupling between the transplanted cells or between the transplanted and host cells with consequent arrhythmias | 30 |
| 6. The use of LVEF for assessing the effects of cell therapy, as this is a load-dependent variable and loss of contractility may be compensated by increases in preload and decrease in afterload that determine changes of the Frank-Starling forces | 31 |
| 7. Incorrect target population of not very sick patients with baseline LVEF ∼50%, with generally a favourable outcome | 31 |
| 8. Existence of well-established alternative therapeutic strategies (PCI, FL, ACE-, and β-blockers) that might mask potential of cell therapy effects | 32 |
| 9. The lack of experimental validation (prove of efficacy) of cell preparation within the trial course |
| Drawbacks | References |
|---|---|
| 1. Low engraftment of BMCs and blood-derived EPCs | 12,24–26 |
| 2. Poor survival of transplanted cells in ischaemic tissue | 27 |
| 3. Failure of adult stem cells to differentiate efficiently into mature and functional cardiomyocytes | 28 |
| 4. Inadequate recruitment of circulating or resident CSCs | 2,29 |
| 5. Anomalous electromechanical coupling between the transplanted cells or between the transplanted and host cells with consequent arrhythmias | 30 |
| 6. The use of LVEF for assessing the effects of cell therapy, as this is a load-dependent variable and loss of contractility may be compensated by increases in preload and decrease in afterload that determine changes of the Frank-Starling forces | 31 |
| 7. Incorrect target population of not very sick patients with baseline LVEF ∼50%, with generally a favourable outcome | 31 |
| 8. Existence of well-established alternative therapeutic strategies (PCI, FL, ACE-, and β-blockers) that might mask potential of cell therapy effects | 32 |
| 9. The lack of experimental validation (prove of efficacy) of cell preparation within the trial course |
BMCs, bone marrow cells; EPCs, endothelial progenitor cells; CSCs, cardiac stem cells; LVEF, left ventricle ejection fraction; PCI, percutaneous coronary intervention; FL, fibrinolysis; ACE-, angiotensin-converting enzyme inhibition.
As a consequence, several strategies have been developed to improve cardiac function in response to cell delivery further. The different strategies and protocols, collectively referred to as ‘cell enhancement’, are discussed in the section “Critical issues on protocols for cell-based therapy”.
In this Position Paper of the ESC WG Cellular Biology of the Heart, we critically review the current approaches using stem cell or cell-based therapies to treat IHD and HF, and discuss promising new strategies for stem cell therapy enhancement, with the aim of increasing the efficacy and outcome of stem cell therapies in the future. The overall objective of this Position Paper is to provide recommendations on how to improve cell-based therapies for cardiac regeneration and repair in IHD and related HF.
Cell sources used in clinical trials
Several types of cells have been used in clinical trials, most of them derived from bone marrow,12,14,15,17–22,33–37 or peripheral blood,38,39 although some studies have used MSCs, cultured from a variety of tissue sources (Table 2). These heterogeneous cell populations used in the early years of regenerative cardiac medicine, have been called “first-generation” stem cells, in contrast to contemporary “second-generation” counterparts. The latter consist of more purified cell populations with a presumed greater potential for cardiac repair and are often derived from non-bone marrow sources, or subjected to genetic and pharmacological “priming” in vitro to enhance their engraftment, survival, plasticity, and paracrine activity. Mesenchymal stem cells exhibit low immunogenicity, making allogeneic application feasible. Since the quality and number of cells may diminish in patients who are older or have comorbidities or genetic defects (reviewed in63), allogeneic MSCs can be used from young healthy individuals. Five systematic reviews and meta-analyses have reported a significant improvement in left ventricle ejection fraction (LVEF) of 2–4% and a reduction in infarct scar size and left ventricular end-systolic volume after intramyocardial transplantation of bone marrow cells.23,31,64–66 To put LVEF into the correct perspective, one must realize that the size of improvement in LVEF determined by cell therapy is comparable, if not higher than what was registered in clinical trials for evaluation of other established therapies for HF, such as angiotensin receptor blockers, aldosterone antagonists, β-blockers, and cardiac resynchronization therapy.67–70 In fact, as summarized in a recent meta-analysis that quantitatively assessed the short-term (4–6 months) therapy-induced changes in LVEF in patients with HF due to left ventricular systolic dysfunction,68 the mean increase in LVEF after subtraction of placebo was 1.3% for angiotensin receptor blockers (valsartan in the Val-Heft trial),67 2.0% for aldosterone antagonists,69 2.7% for cardiac resynchronization therapy,68 and 2.9% for β-blockers (carvedilol).70 Nevertheless, all these therapies are well established to improve clinical outcome in chronic HF. However, biological activity of a cellular product may differ greatly depending on cell source, cell preparation, and cell administration techniques. Therefore, results from meta-analysis should be interpreted with caution, especially in the field of regenerative medicine. Putting together all different trials into one basket becomes more than questionable.
Cell source for therapeutic cardiac regeneration
| Cell type | Name | Sources/origin | Commonly used markers | Advantages/Therapeutic considerations | Disadvantages | References |
|---|---|---|---|---|---|---|
| First generation | Bone marrow and peripheral blood PCs | Bone marrow Peripheral blood | CD117+, CD34+ | Limited regenerative potential Clinical trial phase 3 Minimal improved cardiac function, Limited engraftment Readily cryopreserved Readily genetically manipulated Safety profile Easy accessibility Lack of ethical or immunological problems Extensive clinical experience | Limited differentiation potential Limited yield depending on source | 12,14,15,17–22, 33–35,37–39 |
| Mesenchymal SCs | Foetal origins (Wharton's jelly and cord blood) Adult tissues (i.e. bone marrow or adipose tissue) Developing tooth bud of the mandibular third molar | CD105+, CD117+ | Limited regenerative potential Clinical trial phase 3 Minimal improved cardiac function, Limited engraftment Readily cryopreserved Readily genetically manipulated Source of paracrine factors Easy accessibility Safety profile | Limited differentiation potential Limited yield depending on source | 40 | |
| Side population cells | Cardiac biopsy | Abcg2+, Mdr1+ | Limited regenerative potential No current clinical strategy | Limited yield depending on source | 41–43 | |
| Epicardial PCs | Developing heart | Wt1+, Tbx18+, CD90+, CD44+ | Limited regenerative potential No current clinical strategy | Limited yield | 44–47 | |
| Isl+ PCs | Developing heart | Isl1+ | Limited regenerative potential No current clinical strategy | Limited yield | 48–50 | |
| Second generation | Cells from cardiospheres | Cardiac biopsy | c-kit+, Sca-1+ CD105+, CD29+, CD45− | High regenerative potential Clinical trials phases 1 and 2 Improved cardiac function Limited engraftment | Limited yield | 3 |
| c-kit | Cardiac biopsy | c-kit+, CD45− | High regenerative potential Clinical trials phase 2 Improved cardiac function Limited engraftment | Limited yield The paper describing phase 1 SCIPIO trial received Expression from journal editors at The Lancet, and is still under scrutiny | 2,51–54 | |
| Sca-1 | Cardiac biopsy | ckit+, CD31+, CD14−, CD34−, CD105+, CD45− | High regenerative potential Preclinical stage Limited engraftment | Limited yield | 55–57 | |
| Embryonic SCs | Inner mass of blastocyst | Oct4+, Nanog+, SSEA4+ | High regenerative potential Preclinical stage FIM phase 1 | Ethically controversial source Teratoma formation Immunogenicity | 58 | |
| Induced PSCs | Derived from somatic cells using reprogramming technologies | Oct4+, Nanog+, SSEA4+ | High regenerative potential Preclinical stage Improved cardiomyocyte differentiation | Tumorigenicity | 59–62 |
| Cell type | Name | Sources/origin | Commonly used markers | Advantages/Therapeutic considerations | Disadvantages | References |
|---|---|---|---|---|---|---|
| First generation | Bone marrow and peripheral blood PCs | Bone marrow Peripheral blood | CD117+, CD34+ | Limited regenerative potential Clinical trial phase 3 Minimal improved cardiac function, Limited engraftment Readily cryopreserved Readily genetically manipulated Safety profile Easy accessibility Lack of ethical or immunological problems Extensive clinical experience | Limited differentiation potential Limited yield depending on source | 12,14,15,17–22, 33–35,37–39 |
| Mesenchymal SCs | Foetal origins (Wharton's jelly and cord blood) Adult tissues (i.e. bone marrow or adipose tissue) Developing tooth bud of the mandibular third molar | CD105+, CD117+ | Limited regenerative potential Clinical trial phase 3 Minimal improved cardiac function, Limited engraftment Readily cryopreserved Readily genetically manipulated Source of paracrine factors Easy accessibility Safety profile | Limited differentiation potential Limited yield depending on source | 40 | |
| Side population cells | Cardiac biopsy | Abcg2+, Mdr1+ | Limited regenerative potential No current clinical strategy | Limited yield depending on source | 41–43 | |
| Epicardial PCs | Developing heart | Wt1+, Tbx18+, CD90+, CD44+ | Limited regenerative potential No current clinical strategy | Limited yield | 44–47 | |
| Isl+ PCs | Developing heart | Isl1+ | Limited regenerative potential No current clinical strategy | Limited yield | 48–50 | |
| Second generation | Cells from cardiospheres | Cardiac biopsy | c-kit+, Sca-1+ CD105+, CD29+, CD45− | High regenerative potential Clinical trials phases 1 and 2 Improved cardiac function Limited engraftment | Limited yield | 3 |
| c-kit | Cardiac biopsy | c-kit+, CD45− | High regenerative potential Clinical trials phase 2 Improved cardiac function Limited engraftment | Limited yield The paper describing phase 1 SCIPIO trial received Expression from journal editors at The Lancet, and is still under scrutiny | 2,51–54 | |
| Sca-1 | Cardiac biopsy | ckit+, CD31+, CD14−, CD34−, CD105+, CD45− | High regenerative potential Preclinical stage Limited engraftment | Limited yield | 55–57 | |
| Embryonic SCs | Inner mass of blastocyst | Oct4+, Nanog+, SSEA4+ | High regenerative potential Preclinical stage FIM phase 1 | Ethically controversial source Teratoma formation Immunogenicity | 58 | |
| Induced PSCs | Derived from somatic cells using reprogramming technologies | Oct4+, Nanog+, SSEA4+ | High regenerative potential Preclinical stage Improved cardiomyocyte differentiation | Tumorigenicity | 59–62 |
SCs, stem cells, PSCs, pluripotent stem cells, PCs, progenitor cells.
Cell source for therapeutic cardiac regeneration
| Cell type | Name | Sources/origin | Commonly used markers | Advantages/Therapeutic considerations | Disadvantages | References |
|---|---|---|---|---|---|---|
| First generation | Bone marrow and peripheral blood PCs | Bone marrow Peripheral blood | CD117+, CD34+ | Limited regenerative potential Clinical trial phase 3 Minimal improved cardiac function, Limited engraftment Readily cryopreserved Readily genetically manipulated Safety profile Easy accessibility Lack of ethical or immunological problems Extensive clinical experience | Limited differentiation potential Limited yield depending on source | 12,14,15,17–22, 33–35,37–39 |
| Mesenchymal SCs | Foetal origins (Wharton's jelly and cord blood) Adult tissues (i.e. bone marrow or adipose tissue) Developing tooth bud of the mandibular third molar | CD105+, CD117+ | Limited regenerative potential Clinical trial phase 3 Minimal improved cardiac function, Limited engraftment Readily cryopreserved Readily genetically manipulated Source of paracrine factors Easy accessibility Safety profile | Limited differentiation potential Limited yield depending on source | 40 | |
| Side population cells | Cardiac biopsy | Abcg2+, Mdr1+ | Limited regenerative potential No current clinical strategy | Limited yield depending on source | 41–43 | |
| Epicardial PCs | Developing heart | Wt1+, Tbx18+, CD90+, CD44+ | Limited regenerative potential No current clinical strategy | Limited yield | 44–47 | |
| Isl+ PCs | Developing heart | Isl1+ | Limited regenerative potential No current clinical strategy | Limited yield | 48–50 | |
| Second generation | Cells from cardiospheres | Cardiac biopsy | c-kit+, Sca-1+ CD105+, CD29+, CD45− | High regenerative potential Clinical trials phases 1 and 2 Improved cardiac function Limited engraftment | Limited yield | 3 |
| c-kit | Cardiac biopsy | c-kit+, CD45− | High regenerative potential Clinical trials phase 2 Improved cardiac function Limited engraftment | Limited yield The paper describing phase 1 SCIPIO trial received Expression from journal editors at The Lancet, and is still under scrutiny | 2,51–54 | |
| Sca-1 | Cardiac biopsy | ckit+, CD31+, CD14−, CD34−, CD105+, CD45− | High regenerative potential Preclinical stage Limited engraftment | Limited yield | 55–57 | |
| Embryonic SCs | Inner mass of blastocyst | Oct4+, Nanog+, SSEA4+ | High regenerative potential Preclinical stage FIM phase 1 | Ethically controversial source Teratoma formation Immunogenicity | 58 | |
| Induced PSCs | Derived from somatic cells using reprogramming technologies | Oct4+, Nanog+, SSEA4+ | High regenerative potential Preclinical stage Improved cardiomyocyte differentiation | Tumorigenicity | 59–62 |
| Cell type | Name | Sources/origin | Commonly used markers | Advantages/Therapeutic considerations | Disadvantages | References |
|---|---|---|---|---|---|---|
| First generation | Bone marrow and peripheral blood PCs | Bone marrow Peripheral blood | CD117+, CD34+ | Limited regenerative potential Clinical trial phase 3 Minimal improved cardiac function, Limited engraftment Readily cryopreserved Readily genetically manipulated Safety profile Easy accessibility Lack of ethical or immunological problems Extensive clinical experience | Limited differentiation potential Limited yield depending on source | 12,14,15,17–22, 33–35,37–39 |
| Mesenchymal SCs | Foetal origins (Wharton's jelly and cord blood) Adult tissues (i.e. bone marrow or adipose tissue) Developing tooth bud of the mandibular third molar | CD105+, CD117+ | Limited regenerative potential Clinical trial phase 3 Minimal improved cardiac function, Limited engraftment Readily cryopreserved Readily genetically manipulated Source of paracrine factors Easy accessibility Safety profile | Limited differentiation potential Limited yield depending on source | 40 | |
| Side population cells | Cardiac biopsy | Abcg2+, Mdr1+ | Limited regenerative potential No current clinical strategy | Limited yield depending on source | 41–43 | |
| Epicardial PCs | Developing heart | Wt1+, Tbx18+, CD90+, CD44+ | Limited regenerative potential No current clinical strategy | Limited yield | 44–47 | |
| Isl+ PCs | Developing heart | Isl1+ | Limited regenerative potential No current clinical strategy | Limited yield | 48–50 | |
| Second generation | Cells from cardiospheres | Cardiac biopsy | c-kit+, Sca-1+ CD105+, CD29+, CD45− | High regenerative potential Clinical trials phases 1 and 2 Improved cardiac function Limited engraftment | Limited yield | 3 |
| c-kit | Cardiac biopsy | c-kit+, CD45− | High regenerative potential Clinical trials phase 2 Improved cardiac function Limited engraftment | Limited yield The paper describing phase 1 SCIPIO trial received Expression from journal editors at The Lancet, and is still under scrutiny | 2,51–54 | |
| Sca-1 | Cardiac biopsy | ckit+, CD31+, CD14−, CD34−, CD105+, CD45− | High regenerative potential Preclinical stage Limited engraftment | Limited yield | 55–57 | |
| Embryonic SCs | Inner mass of blastocyst | Oct4+, Nanog+, SSEA4+ | High regenerative potential Preclinical stage FIM phase 1 | Ethically controversial source Teratoma formation Immunogenicity | 58 | |
| Induced PSCs | Derived from somatic cells using reprogramming technologies | Oct4+, Nanog+, SSEA4+ | High regenerative potential Preclinical stage Improved cardiomyocyte differentiation | Tumorigenicity | 59–62 |
SCs, stem cells, PSCs, pluripotent stem cells, PCs, progenitor cells.
Moreover, functional and structural parameters such as LVEF, left ventricular end-systolic volume, and infarct scar size are regarded as surrogate endpoints that cannot substitute hard clinical endpoints.23,31,64–66 Among various possibilities (discussed in Table 1), these modest improvements and the variability between trials have been attributed to the different isolation protocols used, which may profoundly impact the function and number of bone marrow cells or blood-derived EPCs actually delivered to the patient.71,72 Therefore, the general consensus is that assessing cell number and viability along with careful cell characterization and functionality is necessary before delivering cells into patients in any clinical trials. Moreover, the effect of bone marrow mononuclear cells on the incidence of death, recurrent myocardial infarction or stroke and hospitalization for HF remains to be determined in adequately powered prospective clinical trials.
Cardiac-derived progenitor or stem cells have very recently entered the clinical trial arena. Although isolation of these cells from the heart is more invasive than bone marrow, long culture periods are required to obtain sufficient numbers for transplantation, and cell number and functional activity may decline with age, their reported intrinsic paracrine activity is expected to make them potentially good candidates for enhancing myocardial function in HF patients.73–75 Except for the small-scale transendocardial (TE) MSCs and mononuclear bone marrow cells for ischaemic cardiomyopathy trial, comparative clinical data between bone marrow-derived cells (BMCs), MSCs, and CPCs/CSCs is not available in HF patients. A few comparisons have been done in animal models of myocardial infarction (reviewed in76), and MSCs seemed to transfer more benefit on systolic function than BMCs in a chronic large animal model of myocardial infarction.77 Preclinical research thus far suggests the greatest potential functional benefit for CPCs/CSCs from the heart, followed by MSCs, with BMCs having more modest effects on LVEF.78 Conclusions about the effect on mortality of BMC therapy after acute myocardial infarction (AMI) are expected to derive from the ongoing phase III BAMI trial, despite the lack of an placebo control injected group (https://clinicaltrials.gov/ct2/show/NCT01569178).
Of note, there is still no consensus on whether transplanted cell numbers or survival in vivo are crucial for effect size. While trial-based meta-analysis suggested a relationship between cell numbers and effect in clinical trials, individual patient-based meta-analysis have not confirmed this relationship.79
Autologous cells are non-immunogenic and do generally not entail ownership—or ethical issues.80 However, their quality may diminish with age and comorbidities, and genetic defects of the patient will also be present in his/her stem cells and their derivatives. Recent developments now allow the use of allogeneic cells, which can be selected for quality and can be kept ready to use in large quantities “off the shelf” for acute applications.81
Pluripotent stem cells in clinical trials
Another class among the second-generation cells are pluripotent stem cells, both ESCs and iPSCs (Table 2). Prerequisites for clinical application of PSCs for heart regeneration and repair is their efficient and strict differentiation into cardiomyocytes and endothelial cells, which would be directly translated into high effectiveness of the therapy, with minimum risk of undesirable side effects. A clinical trial with ESC-derived cardiomyocytes in severe HF (ESCORT) has been initiated in France and is being monitored with both interest and caution.82,83 Since the same differentiation protocols for ESCs are effective in iPSCs, it may be expected that iPSC-based approaches will also move forward for the treatment of advanced HF. iPSC-derived cardiomyocytes have not yet been tested in humans, largely because of the extra risk of genetic mutation inherent to the reprogramming method as such. The first results of ESC-mediated eye repair are encouraging84 and iPSCs for this aim are in clinical trial since September 2014. The later study is on hold since July 2015 after the identification of a mutation in an oncogene in one of the human iPSC lines (http://www.ipscell.com/2015/07/firstipscstop/). This next-generation iPSC-derived approach is therefore still fraught with uncertainty in the absence of a regulatory framework or guidance about “allowable” levels of mutations and methods of their detection in iPSC products.
Cell-free approaches
A general consensus is that first-generation cells may exert their effects on tissue repair by secretion of paracrine factors. These largely unknown factors may stimulate the myocardium via myocyte salvage, induction of angiogenesis or stimulation of myocyte division. Although the second-generation cells, e.g. CPCs/CSCs and iPSC-derived cardiomyocytes, have been suggested (CSCs) or proven (pluripotent cells) in preclinical trials to have greater regenerative capacity because of their ability to form new myocardium, the contribution of remuscularization vs. “paracrine effects” to overall efficacy has not been demonstrated clinically nor preclinically. Any effects of second-generation cells are thought to be mediated by paracrine mechanisms. Since functional improvement is not necessarily related to cell survival at least in experimental models,85,86 approaches have been developed to mimic the benefit of cell therapy without transplanting the cells. Trialists have attempted to reproduce stem cell-like paracrine effects in ST-elevation myocardial infarction patients using growth factors such as granulocyte-colony stimulating factor (G-CSF).87 All other novel developments in this area are at the preclinical stage. Cell-free strategies include stimulating endogenous repair, e.g. by promoting neovascularization or activating resident progenitor cells.88,89 Mediators of paracrine effects are thought to include growth factors (e.g. erythropoietin, G-CSF),90 episomes,91 and non-coding RNAs,92 mimicking the secretome of donor cells. These factors can also be combined by assembling them in different controlled release formulations, such as microbeads,93 large scaffolds, or injectable biomaterials.94 Recent developments for cell-free approaches that emanate from cells, such as these focus on secreted nano-sized vesicles, called extracellular vesicles, and include microvesicles and exosomes.95,96 These small lipid containing vesicles are capable of transferring proteins, mRNA, and miRNAs between cells, and therefore represent a way for intercellular communication and inducing cardiac repair.97 However, organ selectivity after systemic delivery or inadvertent systemic spread after IC or intramyocardial delivery of these nanoparticles remains unknown and a topic for further scrutiny.
In summary, although the superiority in cardiac repair of one type of cell compared with another has not yet been proven, BMCs continue to be the source of cells most often used in human clinical trials. In cardiac patients, direct comparative data between different cell types is still lacking since adequately powered, randomized clinical trials with head-to-head comparisons of different cell types have not yet been performed. Apart from the risk of immune rejection, which can potentially be circumvented using MSCs, allogeneic somatic/adult cells appear to be safe. To match reported levels of functional cardiac improvement, cell therapy without the cells via paracrine factors may be an interesting alternative. For functional improvement beyond current levels achieved via paracrine actions, new developments will be necessary for proper regeneration of lost tissue.
Critical issues on protocols for cell-based therapy
One major problem for cell therapy is the relatively poor levels of cell retention in the transplanted area, and this may not be limited to first-generation cells, but apply to all cell sources. In fact, ≤10% of injected cells remain at the targeted location. No cells survive when injected into the infarct scar, short-term engraftment is ∼8% regardless of injected cell dose in remote normal myocardium, and in the infarct border zone, the percent survival at 24 h decreases progressively from ∼8% to <1%.27,98
Strategies to improve cell coupling, differentiation, survival, and retention by target area preparation, cell modification, conjugation with biomaterials and/or tissue engineering, and cytoprotection pathways
To improve cell retention, several biomaterial-based approaches have been explored (e.g. hydrogels, cell sheets, prefabricated matrices, microspheres, and injectable nanomatrix).94,99–102 Decellularized extracellular matrices have been shown to promote the healing process via modulation of the host immune response and resistance to bacterial infections.103 An alternative approach, explored in animal models, is the implantation of engineered heart tissue made in vitro from cardiomyocytes and hydrogel.104 Another method is the use of bispecific antibodies that bind to the cells and recognize a cardiac-specific antigen that is only present in injured myocardium.105 Finally, homing can be improved by priming the target organ or tissue with specific treatments, such as extracorporeal shockwaves.106 Localized hypoxia, inflammation, excessive oxidative stress, lack of supporting cells, poor supply of nutrients, and fibrosis promote apoptosis or necrosis of the grafted cells. Thus, the efficiency of cell therapies might be improved by using genetic engineering tools, including overexpression of pro-survival genes (e.g. Akt, Pim-1 kinase, ERK1/2, HIF-1α, haeme-oxygenase 1, GATA4, heat shock protein 27, miRNA-1, myocardin, and protein kinase G1α) or angiogenesis-initiating genes (e.g. VEGF, MYDGF, fibroblast growth factor (FGF)-2, SDF-1, and PDGF) in the cells to be transplanted or by transplanting the cells together with pro-survival or pro-angiogenic factors.76,98,107–113 Interestingly, exposure of cells to sub-lethal hypoxia increased the tolerance of these cells to the harsh environment after transplantation.114 These preconditioned cells also showed increased differentiation, enhanced paracrine effects leading to increased trophic support, and improved homing to the lesion site.114 Transplantation of preconditioned cells helped to suppress inflammatory factors and immune responses, and promoted heart function.114 In addition, transient modulation of cell specification towards myogenic differentiation, e.g. via microRNAs, could also be beneficial in increasing the amount of myocardium. miR-1 and -499 are excellent candidates as they can enhance both differentiation in vitro95 and in vivo.115 Another approach to promote transplanted cell survival is to modulate the inflammatory environment (using TSG-6, IL-1 inhibitor).109,116 Finally, a significant barrier to the therapeutic use of most cell populations with the exception of ESCs and iPSCs, is their limited cardiac differentiation potential despite the use of “cardiogenic cocktails” (containing TGF-β1, BMP-4, activin A, retinoic acid, IGF-1, FGF-2, α-thrombin, and IL-6) and overexpression of cardiac transcription factors.117–119 In addition, these stem cells fail to electromechanically integrate.120 This limitation has been partially solved by overexpressing the two key proteins, N-cadherin, and connexin 43, but clinical translation remains to be fully investigated. In contrast, human PSCs can now be routinely differentiated with high efficiency (>80%) into cardiomyocytes.121 However, the cardiomyocyte populations may contain varying proportions of atrial, ventricular, and nodal cardiomyocytes.122,123 This is a critical issue as they have unique mechanical and electrical properties and thus, the implantation of a mixture of these cells harbors the risk of arrhythmias.124 In addition, all of these cardiomyocyte types are immature and beat spontaneously, another source of arrhythmogenic risk. Consequently, even though many protocols primarily give rise to ventricular-like cardiomyocytes, it is important to refine the differentiation protocols to produce pure populations of defined cardiomyocyte phenotype.125,126 In addition, robust irreversible cardiac lineage differentiation of all transplanted cells is critically important to avoid the formation of teratomas.
Stem cell rejuvenation
Aging or comorbidities may cause a reduction in the number and function of tissue-resident and circulating cells.63,127,128 Several proteins and signaling pathways have been identified that are capable of reverting the process of cell senescence, including Pim-1 kinase,121–124 NOTCH1,129–131 telomerase, and myocardin.132 Pim-1 kinase has anti-senescence and anti-apoptotic effects in CSCs as well as in MSCs.133 Activation of the NOTCH1 signaling pathway results in remarkable rejuvenation of satellite muscle cells associated with enhanced proliferation, increased telomere lengths, and decreased susceptibility to replicative senescence.129 The overexpression of telomerase and myocardin genes increases cell survival, proliferation, cardiomyogenic,132,134 and smooth muscle differentiation in vitro.135 After overexpression of genes encoding for “rejuvenating factors’ and in vitro expansion, genetically modified cells may secrete high amounts of the “regenerating factor”, either transiently or permanently, at the transplantation site.107,136 Most of the approaches for genetic modification of cells requires cell manipulation with some risk of cell contamination, accumulation of mutations during in vitro culture, or insertional activation of other genes, due to the use of viral vectors. Taken together, the design of new protocols for aged cell rejuvenation would allow improved cell preparation and clinical application of cells in older patient populations, taking into account the safety problem of the genetic manipulation of cells.
Enhancing endogenous cardiac regeneration
Recent studies have demonstrated that cardiomyocyte turnover occurs throughout life in mammals, including humans.137–141 While the estimated rate of human cardiomyocyte renewal is controversial, most studies report an annual turnover rate of 1%, which increases after injury. However, the intrinsic capability in humans to regenerate injured myocardium after massive ischaemic cell death is too low to be of functional relevance. It has been suggested that transplanted cells may exert their beneficial effects by secreting cytokines and growth factors promoting cardiomyocyte proliferation, recruitment and activation of CPCs, induction of vessel formation, reduction of fibrotic scars, and inhibition of apoptosis.142 In addition, modulation of macrophage and regulatory T-cell function can improve healing, repair, and regeneration.143,144 Another approach to enhance endogenous cardiac repair is the induction of cardiomyocyte proliferation, a mechanism described in neonatal mice, zebrafish and newts in response to injury,145 although never in adult mammals. However, cardiomyocyte regeneration after myocardial infarction may be promoted also by administration of FGF-1, p38 MAP kinase inhibitor,146 blocking the Hippo pathway or upregulating the downstream Hippo effector Yes-associated protein (Yap),147 activation of Erb-B2 Receptor Tyrosine Kinase 2 signaling, or application of the human Fstl1 protein via an epicardial patch.148 Yet, so far it is unclear whether the observed functional improvement is due to cardiomyocyte proliferation and generation of new cardiomyocytes.149
Recently, promising new directions have been explored that are promoting for in situ regeneration, in which cellular transplantation will become redundant. Hereby, fibroblasts are being directly converted into cardiomyocyte-like cells by using a combination of three cardiac developmental transcription factors, Gata4, Mef2c, and Tbx5.150 These reprogrammed cells express several cardiac-specific markers and exhibit spontaneous contractions. Still in its infancy and efficiency being low, this approach will potentially allow the conversion of extracelluar matrix producing cells—the myofibroblasts in the scarred area—into cardiomyocytes in vivo.151 Follow-up studies are exploring the use of different combinations of transcription factors,152,153 or efficiencies being improved via microRNA-mediated reprogramming.154
Cell-tracking systems
In vivo cell tracking involves either ‘direct’ physical labelling of cells by incubating them with a contrast agent, or ‘indirect’ genetic labelling by transfecting cells with a reporter gene construct. The position of, and signal from these labels can then be tracked using various imaging modalities, including clinical scanners, such as positron emission tomography (PET), single photon emission computed tomography (SPECT) and magnetic resonance imaging (MRI) (reviewed in155,156). All imaging modalities can provide information regarding short-term kinetics of transplanted cells and their effects on cardiac function, but are not capable to assess the long-term engraftment. Given its high anatomical resolution and safety profile allowing serial longitudinal evaluations, MRI has been commonly used to track cells in clinical trials.157 However, MRI might detect macrophages that ingest the marker after the cell (derivative) had lysed. Other limitation could be the loss of signals over the time related to cell death or cell division. Safety concerns regarding the effects of genetic manipulation of cells currently limit the use of genetically modified cells in clinical trials, and thus long-term cell tracking. However, combination approaches relying on the simultaneous co-registration of different imaging modalities (nuclear medicine combined with CT or MRI) might overcome the limitations of individual imaging techniques, and represent powerful tools to gain insight into the delivery, engraftment, survival, off-target, and possible adverse effects of transplanted stem and progenitor cells. Given the indispensable role of cell tracking in clinical trials, the feasibility of imaging should be included in preliminary proof of concept studies, and considered among inclusion or exclusion criteria, but will limit cell transfer studies to only a few centers that have access to multimodal imaging expertise.
Controls, data reproducibility, standardization issue, and data quality
Over the past few years, concerns have been increasingly voiced about experimental reproducibility across the whole biomedical research fields,158,159 not least in cell therapy. For example, a recent paper searching for errors in published cardiac clinical trials using autologous BMCs reported that the greatest enhancement of LVEF was described in those studies with the most discrepancies or errors in factual reporting.160 The pervasive risk of neglecting basic rules of clinical trial design in stem cell trials has been demonstrated in a recent review.161 On the other hand, phase II studies, where the aim is to prove efficacy should be designed to assess several primary end-points, which might include structural evaluations, cardiovascular physiological measurements, biomarkers, functional capacity, and quality of life.162
The choice of appropriate controls and methodological rigor may be more demanding in the field of cell therapy if, for example, the need for a myocardial biopsy to harvest autologous stem cells complicates double-blinding. A pragmatic alternative is to use a crossover study design, in which each patient is randomly assigned to a sequence of treatments. However, where reagents, such as cytokines are administered in conjunction with cells, a control group with cytokines alone should also be included. Another issue is the choice of the right placebo control, which, in some cell therapy trials, simply consisted of transparent saline solution which can be easily be distinguished visually from serum.
Standardization of cell isolation and processing procedures is highly desirable in order to facilitate comparisons between trials and to enable meta-analyses. Sex of donor-cells should also be clearly stated, as sex differences can affect the regenerative potential of transplanted cells. Finally, standardization of patient populations and stratifications should be attempted. It has been proposed that reference MSCs be developed to facilitate comparison between studies.163
In summary, cell-based therapies would benefit significantly from different protocols collectively referred to as cell enhancement, including possible priming of host tissue with cytokines to increase homing, preconditioning of transplanted cells, drugs and pro-survival factors, genetic engineering of cells, and the use of biomaterials. All of these strategies could contribute to improving cell retention and promote cell survival, proliferation, differentiation, and induction of neo-angiogenesis. Nevertheless, irrespective of cell enhancement, pilot studies to understand where the cells go by choosing the best tracking system in vivo, and adherence to well-established rules for the design of robust clinical trials, are minimum requirements for any cell protocol to assess actual effectiveness of cell-based clinical interventions.
Clinical trial design
Safety and ethical issues
The design of randomized controlled clinical trials that are able to ascertain the long-term safety of cell therapy can be challenging from an ethical perspective, and encompass issues related to:164 (i) public perception of cell therapy—heightened expectations may influence the patient's decision to participate in clinical studies with cell therapy and may also affect the randomization procedure, with a preference to be in the treatment arm of the study rather than in the control group; (ii) conflicts of interest—commercial interests may place pressure on researchers to investigate cell therapies which are not yet ready for clinical testing; (iii) risks vs. potential benefits—given the invasive nature and uncertainties surrounding cell therapy, the potential risks may be difficult to define, thereby making the consent procedure all the more challenging; (iv) choice of study outcome measure—there is a fine balance between choosing a surrogate endpoint which provides mechanistic insight, and clinically relevant endpoints.
Patient selection (co-morbidities and co-medications)
When considering efficacy of cell therapy, a better understanding of cell biology and the interaction between treatment and patient-specific cardiovascular risk factors, co-morbidities (such as age, gender, diabetes, hypertension, dyslipidaemia, smoking, depression, and psychological stress), and routine medications is required. All major co-morbidities and co-medications in patients with IHD are potential confounders of the efficacy of cell therapy, because they affect the quality of source cells as well as the response of host tissue to the transplanted cells.32,165,166 Autologous or allogeneic haematopoietic cell transplantation for haematologic diseases was the first type of cell therapy, the outcome of which was correlated with comorbidity indices.167 However, no data are available on comorbidity index or score systems to be used in clinical cell therapy trials in order to objectively and reproducibly assess the possible interference of pre-existing co-morbidities and co-medications with outcome.165 Recently, micro-array platforms identified global transcriptional and functional differences between CPCs/CSCs and BMCs. These tools can help examine the true functionality of stem cells and customize cellular therapy according to specific patient variability in terms of co-morbidities or age.168 In this regard, key points that should be considered are the following: (i) roughly equal stratification of patients into risk groups; (ii) the inclusion of possible confounders in the analyses of outcomes; (iii) evaluation of aging as a three-dimensional variable incorporating chronologic age (which is a poor predictor of cell therapy outcomes, probably due to a lack of data on organ dysfunctions,169), co-morbidities, physical function, nutritional, and cognitive status; (iv) developing useful prognostic biomarkers and co-morbidity index that could help understanding correlations between co-morbidities with either cell biology and host response before any cell therapy; (v) determining the transcriptional and functional variation of adult stem cells for autologous cell therapy, that could help assessing which cell populations are optimal for diverse aged patients with existing co-morbidities and co-medications.
In summary, careful attention must be given to a variety of factors (including age, gender, co-morbidities, concomitant medications, and any other cardiovascular risk factors) that may interfere with the regenerative potential of cell therapy in the setting of IHD and HF. The development of useful prognostic biomarkers and co-morbidity indexes could help to objectively assess the weight of these factors in both preclinical and clinical trials.
Clinically relevant delivery routes, cell dose, and timing of delivery
Catheter-based IC cell infusion using a perfusion balloon catheter during stop flow conditions is the mostly used delivery route in clinical trials, with the following drawbacks: (i) the potential non-selective distribution pattern of the transferred cells, with exclusion of infarcted and border area in the case of an occluded coronary artery; (ii) the need for the cells to transmigrate from the vessel lumen into the myocardium; (iii) the possible occurrence of microembolisms with subsequent myocardial dysfunction. Intravenous administration is limited by entrapment of the donor cells in the capillaries of the lungs. Direct myocardial injection during open chest surgery is the most precise and accurate type of delivery. Transcatheter TE cell injection through the femoral artery and the aortic valve is less invasive.
Direct comparison of IC and TE using electromechanical mapping guidance and surgical delivery of autologous Indium-oxine-labelled bone marrow-derived MSCs was studied in a chronic pig model of ischaemic cardiomyopathy, however, did not show any significant difference in cell delivery efficiency to the myocardium nor in differences in safety profile.170
Recently, retrograde bone marrow cell injection via coronary sinus infusion has been tested as safe and feasible in patients with either ischaemic or non-ischaemic HF.171
With respect to cell dose (reviewed in76), it should be noted that in the vast majority of pre-clinical and clinical studies, dosing has been non-systematic and empirically assessed, guided more by feasibility and accessibility rather than by intentional dosage optimization. This has contributed to the still open question of how many cells should be delivered in order to achieve clinical benefit. Although a dose–response curve is difficult to obtain when using autologous cells from individual patients, the relative number of cells with high functional activity, rather than the absolute number of cells, should be considered to describe the dose response. Mean absolute numbers of cells infused into the coronary circulation of patients with IHD and HF range from 1.2 × 107 to 2.05 ± 110 × 108 bone marrow cells and from 1 × 106 to 25 × 106 CSCs (reviewed in76).
The optimal timing of donor cell delivery also remains debated. Although no consensus has been reached, between 4169 and 8 days172 after AMI onset seemed to be the optimal time point for BMCs or circulating blood-derived progenitor cells delivery into an infarct-related coronary artery, based on limited homing studies and the time course of inflammatory responses after myocardial infarction.
How to assess the clinical benefit of cell therapy (including follow-up)
In the vast majority of trials, the primary endpoint has been the evaluation of left ventricular size and global systolic function before and after treatment (reviewed173). Small (if any) improvements of LVEF have been observed in cell-treated patients by two-dimensional echocardiography, MRI, left ventricular angiography, or radionuclide ventriculography performed at different time points and with different acquisition and analysis protocols (reviewed in173). Given the controversial outcomes of previous clinical trials, future studies should avoid imaging methodologies with poor reproducibility, should standardize timing of image acquisition and analysis protocols, and more comprehensively evaluate the potential benefits deriving from cell therapy. Indeed, implementation and standardization of other techniques, such as 3D echocardiography,174 strain/strain rates,175,176 tissue Doppler echocardiography,177,178 and MRI might be extremely helpful to identify more sensitive markers of cardiac improvement. It is important to emphasize that, at the present time, MRI currently provides the most accurate, comprehensive, and reproducible measurements of cardiac chamber dimensions, volumes, function, and infarct size compared with other techniques,179,180 and therefore should be performed in cell-treated patients enrolled in clinical trials whenever possible at baseline, after treatment, and during follow-up. In addition to MRI, myocardial viability should be determined by 18F-FDG PET assessing glucose metabolism, alone or in association with dobutamine stress echocardiography, since all studies using 18F-FDG have shown an improvement in myocardial viability,181,182 but this beneficial effect has not always been paralleled by an increase in contractile reserve.183 Furthermore, possible limitation of 18F-FDG PET is the radiation exposure of patients undergoing serial imaging. Finally, to precisely determine the effects of cell therapies on vasculogenesis, serial quantitative PET evaluations of global, and regional myocardial perfusion might be extremely valuable.20,182,184 Independent of the specific technology, centralized evaluation by independent, and blinded core labs should be standard.
In addition to the above endpoints, real, clinically relevant endpoints should also be used in future clinical trials, as, e.g. indicated in the BAMI trial that is focused on the effect of IC reinfusion of BMCs on all-cause mortality in AMI (NCT01569178). Although such trials need enough power and are costly, they are essential to demonstrate the net clinical benefit for patients. Additional standard tests that should be considered, include quality of life assessment, number of hospitalizations, 6 min walk tests, and death over several years' follow-up.
In summary, how patient selection takes place, what the best clinically relevant delivery routes are for cell administration, which cell dose and timing of delivery should be used and what clinical endpoint should be analyzed and by which method, are the most crucial aspects in clinical trials investigating the effects of cell therapy. Adequately powered large-scale clinical trials, taking into account all the possible safety and ethical issues, considering cell function as one of major predictors of successful cell therapy, and focusing on hard clinically meaningful endpoints, are mandatory to determine whether the observed functional improvement reported in some studies can be extended to others and indeed translates into increased survival and reduced morbidity.
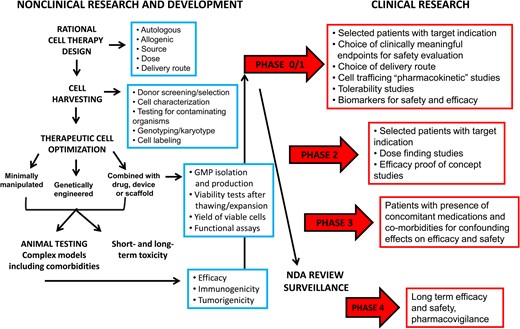
Flowchart of experimental design starting from preclinical studies and ending to the human clinical trials.
Recommendations
In Figure 1, we provide a flowchart of experimental design starting from non-clinical studies and ending with the human clinical trials. To this translational pathway, we would like to make the following recommendations when assessing the clinical potential of conventional cell-based therapy, as well as novel strategies of cell enhancement for cardiac regeneration and repair in IHD and HF patients:
Conventional cell-based therapy have reported efficacy and safety in most experimental myocardial infarction models tested, including those in large animals, but in human clinical trials in IHD and HF patients, only safety of cell therapies has been shown. Therefore, future pre-clinical studies using cell-based therapies should be designed to address specific hypotheses on modes of delivery and mechanisms of efficacy, rather than safety and efficacy endpoints only;
Based on the expected clinical trial outcome, a careful selection of cell source is essential. Whereas first-generation cells might be useful for stimulation of endogenous repair mechanisms or angiogenic effects, second-generation cells truly aim at replacing damaged myocardium. A comparison of different cell types, or a combination of cell types in randomized clinical trials has not yet been performed, but are being planned in future trials of chronic ischaemic HF.
Assessing cell number and viability along with full cell characterization including cell function, should be done in every clinical trial;
Poor cell retention remains a major issue. To further boost both cellular and paracrine effects, effective carrier materials and/or engineering approaches or pre-treatment strategies of cells or target tissues should be further developed.
To maximize successful translation of novel cell enhancement strategies, it is of primary importance to ensure that the efficacy of preclinical studies is validated whenever possible in the presence of confounding factors, such as age and gender and common cardiovascular co-morbidities as well as their routine medications. Likewise, for clinical application of genetically manipulated cells, it is important to ensure the safety of the injected cells in terms of genetically and epigenetically stability, efficient and reproducible differentiation, and highly reliable cell purification;
The usage of hard clinically meaningful endpoints is mandatory to determine whether functional improvement indeed translates into increased survival and reduced morbidity.
Conclusions
The early promise of cell therapy has not yet been fulfilled. First-generation cells and their secretomes that aim at myocardial salvage and stimulating the endogenous repair mechanisms of the heart through pro-angiogenic or prosurvival activity should be carefully selected depending on the desired effect. Second-generation cells such as pluripotent stem cells are indisputably capable of forming beating contractile cardiomyocytes, but large surviving grafts of injected cells are rarely observed.185 Combining these cell types with biomaterials may enhance the outcome of present cardiac cell transplantation therapy, by truly replacing the damaged myocardium with muscular grafts. Other strategies to empower the donor cells, referred to as cell enhancement, may further stimulate paracrine effects, but new developments will be necessary to achieve cardiac regeneration, e.g. by stimulating endogenous cardiac regeneration. Moreover, the selection of appropriate clinical endpoints, patient population, and delivery strategies are crucial aspects to understand the clinical effects. Furthermore, focusing on hard clinical endpoints in future cell-based trials is mandatory to determine whether any observed functional improvement translates into increased survival and reduced morbidity.
Funding
This work was supported by Cariplo (Rif. 2011-0566) and the Italian Ministry of Research (PRIN 2010YK7Z5K_007) (to R.M. and Prof. Raffaele De Caterina); by Veni (ZonMW) 91612147 and Netherlands Heart Foundation 2013T056 (to L.W.V.L.); by the NIHR Biomedical Research Centre (BRC233/CM/SD/101320), British Heart Foundation (PG/15/52/31598), and Medical Research Council (MR/K002066/1) (to S.M.D.); by the Italian Ministry of Health (GR-2009-1596220) and the Italian Ministry of Research (RBFR124FEN) (to C.P.); by ZonMW (91612147) and Netherlands Heart Foundation (2013T056) (to L.v.L.); by the Emerging Fields Initiative (EFI) for Cell Cycle in Disease and Regeneration (CYDER) from the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) (Germany) (to F.B.E.); by the German Centre for Cardiovascular Research (DZHK) and the German Ministry of Education and Research (BMBF), the German Research Foundation (DFG Es 88/12-1), the European Research Council (ERC AG IndivuHeart), and the European Commission (FP7 Biodesign) (to T.E.); by the Netherlands CardioVascular Research Initiative (CVON), the Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development, and the Royal Netherlands Academy of Sciences (to J.P.G.S.); by Norwegian Council on Cardiovascular Diseases (to K.Y.), by National Heart, Lung and Blood Institute NHLBI, rif. UM1 HL087365-10 (to J.T.W.), and by H2020-SMEINST-1-2015 “Infarnosys” (to Pharmahungary). P.F. was a Szentágothai Fellow (TÁMOP-4.2.4.A/2-11/1-2012-0001) of the National Excellence Program of Hungary.
Conflict of interest: T.E. and C.L.M. are co-founders of EHT Technologies GmbH, Hamburg and Pluriomics BV, respectively. P.F. is a founder and CEO of Pharmahungary Group.
References
Author notes
These authors contributed equally.


{kind=link}