





Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia in man, causing substantial morbidity and mortality with a major worldwide public health impact. It is increasingly recognized as a highly heritable condition. This study aimed to determine genetic risk factors for early-onset AF.
We sequenced the whole genomes of 8453 Icelanders and imputed genotypes of the 25.5 million sequence variants we discovered into 1799 Icelanders with early-onset AF (diagnosed before 60 years of age) and 337 453 controls. Each sequence variant was tested for association based on multiplicative and recessive inheritance models. We discovered a rare frameshift deletion in the myosin MYL4 gene (c.234delC) that associates with early-onset AF under a recessive mode of inheritance (allelic frequency = 0.58%). We found eight homozygous carriers of the mutation, all of whom had early-onset AF. Six of the homozygotes were diagnosed by the age of 30 and the remaining two in their 50s. Three of the homozygotes had received pacemaker implantations due to sick sinus syndrome, three had suffered an ischemic stroke, and one suffered sudden cardiac death.
Through a population approach we found a loss of function mutation in the myosin gene MYL4 that, in the homozygous state, is completely penetrant for early-onset AF. The finding may provide novel mechanistic insight into the pathophysiology of this complex arrhythmia.
Introduction
Atrial fibrillation (AF) is primarily a disease of the elderly. It has an overall population prevalence of ∼1%, but the prevalence increases markedly with age.1 On the other hand, risk estimates of early-onset AF (diagnosed prior to 60 years of age) are only 3% for males and 1% for females.2 In up to 30% of AF cases no predisposing condition can be identified and the disease has traditionally been classified as lone AF.3
Familial clustering of AF, particularly of early-onset cases, is well recognized. We reported strong heritability among unselected AF cases and showed that first-degree relatives of Icelanders with early-onset AF are nearly five times more likely to develop AF than the general population.4 Mutations in several genes, identified through linkage and candidate gene studies, have been found in both familial and sporadic AF.5 Most of these are ion channel genes.5 In 2007, the first common sequence variants that associate with common AF, close to PITX2, were discovered through genome-wide association6 and several have been found since,5 including a variant in MYH6, encoding the alpha heavy chain subunit of cardiac myosin.7 In this study, we used a large population-based resource of whole-genome sequence data to search for sequence variants that affect the risk of early-onset AF.
Methods
We have briefly described these data previously in a paper on whole-genome sequencing in Icelanders.8 Here we present the findings in full, including detailed phenotypic data. The investigation was approved by the Icelandic Data Protection Authority and the National Bioethics Committee of Iceland. The study complies with the Declaration of Helsinki.
Study populations
The deCODE Icelandic AF population consisted of Icelanders diagnosed with AF (ICD 10 code I48 and ICD 9 code 427.3) according to electronic medical records (EMR) at Landspitali, The National University Hospital in Reykjavik and Akureyri Hospital (the two largest hospitals in Iceland) from 1987 to 2012. This analysis was restricted to 1799 cases diagnosed before age 60 years, comprised of 1183 single nucleotide polymorphism (SNP) chip-typed individuals and 616 close relatives (familial imputation, see Generation of genotype data below, see also Supplementary material online, Figure S1). The control group of 337 453 people was made up of 137 445 chip-typed individuals enroled in various studies at deCODE genetics and 200 008 of their close relatives,8 representing a large fraction of the Iceland population. The EMR diagnoses do not always represent the earliest age at diagnosis but rather an upper bound of the earliest age at diagnosis. Detailed medical record review was performed for the homozygous carriers of the c.234delC deletion, including review of both electronic and paper medical records, electrocardiograms, and echocardiograms. Samples from the Tromsø Study and the Hong Kong Atrial Fibrillation Study were genotyped for the MYL4 c.234delC mutation (Supplementary material online).
Generation of genotype data
We have previously described methods to interrogate whole genomes of large numbers of Icelanders and to search for associations with various traits8 (see also Supplementary material online). In brief, 137 445 Icelandic samples were genotyped with Illumina microarrays (chip-typed), a method to determine the alleles of predefined SNPs for each individual. Most of the chips used contained 600 000 SNPs. The whole genomes of 8453 chip-typed Icelanders were also sequenced using standard TrueSeq methodology (Illumina) to a mean depth of at least 10X (median 32X). This method determines the near-complete DNA sequence for each individual. The sequencing depth (coverage) refers to the average number of reads representing a given nucleotide in the reconstructed sequence. Genotypes of the 25.5 million sequence variants identified through sequencing (SNPs and indels) were then imputed into all chip-typed Icelanders and their close relatives (familial imputation) (Supplementary material online). Imputation refers to the statistical inference of unobserved genotypes using known haplotypes in the population.
Microsatellite genotyping
PCR amplifications were set up and pooled using Zymark SciClone ALH 500 robots. The sequences of the forward and reverse primers used for genotyping were TCACTGGGGATTGGAAGGTA and GGCATT GGT A GGGTTCTGG, respectively. Conditions for PCR reactions are described in the Supplementary material online. The PCR products were supplemented with an internal size standard and the fragments were separated and detected on an Applied Biosystems model 3730 sequencer, using Genescan (v. 3.0) peak-calling software. Alleles were called using an internal allele-calling program.9
Association testing
Logistic regression was used to test for association between SNP genotypes and disease. Disease status was treated as the response and the expected allele counts from imputation as covariates for the multiplicative model. For the recessive model, the product of the maternal and paternal expected minor allele counts was used as a covariate (Supplementary material online).
Inflation factor adjustment
In order to account for relatedness and stratification within the Icelandic case and control samples, we applied the method of genomic control based on chip markers.10 The correction factor based on linkage disequilibrium (LD) score regression was 1.12 for the multiplicative model and 1.05 for the recessive model. All P values were adjusted accordingly.11
Age of the mutation
We estimated the age of the c.234delC mutation by examining the decay of LD between the mutation and neighbouring chip markers.12 These age estimates were then corrected based on the Luria-Delbrück model.13 A single estimate for the age of the mutation in generations was then obtained by taking the geometric mean of the age estimates for each SNP. This was multiplied by 30 to obtain an estimate of the age of the mutation in years.
Results
A single nucleotide deletion in MYL4 is associated with early-onset AF
We found that the two most significant associations with early-onset AF under the multiplicative model were with variants at previously reported loci, PITX26 and ZFHX3.14 No other variants passed the Bonferroni threshold for genome-wide significance (P < 0.05/25.5 million = 2.0 × 10−9, Supplementary material online, Figure S2).
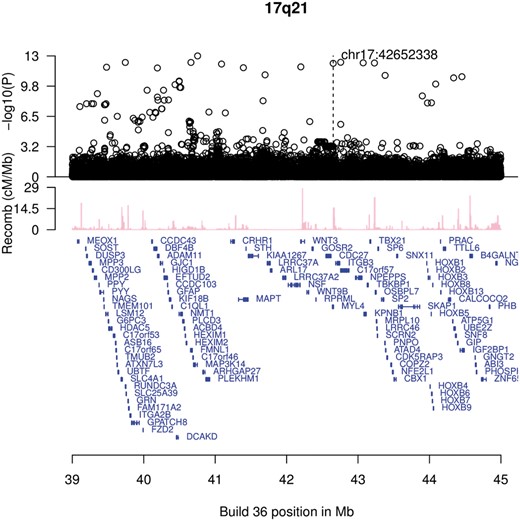
An overview of the 17q21 region around MYL4 showing recessive association with early onset atrial fibrillation. Shown are—log10 P values for association under a recessive mode of inheritance of imputed single nucleotide polymorphisms based on genome-wide sequencing as a function of their build 38 coordinates. All eight homozygous carriers of the c.234delC mutation were counted as cases in the analysis. Genes are shown in blue. Recombination rates are reported in centi-Morgans per megabase (cM/Mb).

A protein sequence alignment of the wild-type and mutated MYL4 gene products illustrating the effect of the c.234delC mutation. Conserved amino acid residues are depicted as a dot (.), the portion of the mutated protein rendered out of frame is within a box and missing residues as a cause of a premature stop codon are depicted as a dash (-). Secondary and tertiary protein structure properties are shown above and below the alignment, respectively (UniProtKB: P12829).
Within the 3.9Mb interval spanned by the sequence variants reaching genome-wide significance, the average coverage was >5X for 99.9% and >10X for 99.4% of exonic positions, sufficient coverage for a sequence variant with 0.58% minor allele frequency (expected 98 sequenced carriers) to be observed with high probability.
The test for association of c.234delC with early-onset AF was based on imputing the c.234delC genotype status into chip-typed individuals using a whole-genome sequenced sample of 8453 Icelanders as the basis for the imputation, 99 of whom were heterozygous for the deletion. The quality of imputation was high (information = 0.95), estimated by a measure of imputation information (see Supplementary material online). To confirm the existence of the c.234delC deletion and to validate its imputation, we performed microsatellite genotyping of 879 Icelanders, including 24 imputed heterozygotes and three imputed homozygotes. We saw no mismatches between imputed and observed genotypes .
The imputation of c.234delC led to the identification of six chip-typed Icelanders who were homozygous carriers. Five of them had been diagnosed with early-onset AF. Familial imputations additionally revealed two sisters who were homozygotes (Table1 and Figure3), one of whom had been diagnosed with AF at age 56.
Clinical information on the eight homozygous carriers of c.234delC in MYL4
| Electrocardiogram | ||||||||
|---|---|---|---|---|---|---|---|---|
| Sex | Current age | Age at discharge diagnosis of AFa | Age at diagnosis of AFb | Ischemic stroke | Permanent pacemaker | PR (ms) [120–220] | QRS (ms) [80–100] | QTc (ms) [370–450] |
| Male | 47 | 25 | 14 | No | Yes at 40 | 195 | 115 | 410 |
| Female | 53 | 27 | 21 | No | No | — | 100 | 390 |
| Male | 58 | 32 | 28 | No | Yes at 41 | 165 | 82 | 405 |
| Female | Died 64c | — | 53 | No | No | — | 98 | 395 |
| Female | Died 58 | 56 | 56 | Yes at 56 | No | 240 | 84 | 400 |
| Male | 57 | 46 | 20 | Yes at 46 | Yes at 47 | — | 110 | 405 |
| Female | 66 | 47 | 30 | No | No | 160 | 98 | 380 |
| Male | 59 | — | 15 | Yes at 35 | No | — | 95 | 390 |
| Electrocardiogram | ||||||||
|---|---|---|---|---|---|---|---|---|
| Sex | Current age | Age at discharge diagnosis of AFa | Age at diagnosis of AFb | Ischemic stroke | Permanent pacemaker | PR (ms) [120–220] | QRS (ms) [80–100] | QTc (ms) [370–450] |
| Male | 47 | 25 | 14 | No | Yes at 40 | 195 | 115 | 410 |
| Female | 53 | 27 | 21 | No | No | — | 100 | 390 |
| Male | 58 | 32 | 28 | No | Yes at 41 | 165 | 82 | 405 |
| Female | Died 64c | — | 53 | No | No | — | 98 | 395 |
| Female | Died 58 | 56 | 56 | Yes at 56 | No | 240 | 84 | 400 |
| Male | 57 | 46 | 20 | Yes at 46 | Yes at 47 | — | 110 | 405 |
| Female | 66 | 47 | 30 | No | No | 160 | 98 | 380 |
| Male | 59 | — | 15 | Yes at 35 | No | — | 95 | 390 |
Shown are the sex, year of birth, and current age.
Age at diagnosis according to hospital electronic discharge diagnosis codes.
Age at diagnosis of AF according to review of medical records, ischemic stroke diagnosis, permanent pacemaker placement and the results of electrocardiogram analysis (normal range in brackets).
Sudden cardiac death.
Clinical information on the eight homozygous carriers of c.234delC in MYL4
| Electrocardiogram | ||||||||
|---|---|---|---|---|---|---|---|---|
| Sex | Current age | Age at discharge diagnosis of AFa | Age at diagnosis of AFb | Ischemic stroke | Permanent pacemaker | PR (ms) [120–220] | QRS (ms) [80–100] | QTc (ms) [370–450] |
| Male | 47 | 25 | 14 | No | Yes at 40 | 195 | 115 | 410 |
| Female | 53 | 27 | 21 | No | No | — | 100 | 390 |
| Male | 58 | 32 | 28 | No | Yes at 41 | 165 | 82 | 405 |
| Female | Died 64c | — | 53 | No | No | — | 98 | 395 |
| Female | Died 58 | 56 | 56 | Yes at 56 | No | 240 | 84 | 400 |
| Male | 57 | 46 | 20 | Yes at 46 | Yes at 47 | — | 110 | 405 |
| Female | 66 | 47 | 30 | No | No | 160 | 98 | 380 |
| Male | 59 | — | 15 | Yes at 35 | No | — | 95 | 390 |
| Electrocardiogram | ||||||||
|---|---|---|---|---|---|---|---|---|
| Sex | Current age | Age at discharge diagnosis of AFa | Age at diagnosis of AFb | Ischemic stroke | Permanent pacemaker | PR (ms) [120–220] | QRS (ms) [80–100] | QTc (ms) [370–450] |
| Male | 47 | 25 | 14 | No | Yes at 40 | 195 | 115 | 410 |
| Female | 53 | 27 | 21 | No | No | — | 100 | 390 |
| Male | 58 | 32 | 28 | No | Yes at 41 | 165 | 82 | 405 |
| Female | Died 64c | — | 53 | No | No | — | 98 | 395 |
| Female | Died 58 | 56 | 56 | Yes at 56 | No | 240 | 84 | 400 |
| Male | 57 | 46 | 20 | Yes at 46 | Yes at 47 | — | 110 | 405 |
| Female | 66 | 47 | 30 | No | No | 160 | 98 | 380 |
| Male | 59 | — | 15 | Yes at 35 | No | — | 95 | 390 |
Shown are the sex, year of birth, and current age.
Age at diagnosis according to hospital electronic discharge diagnosis codes.
Age at diagnosis of AF according to review of medical records, ischemic stroke diagnosis, permanent pacemaker placement and the results of electrocardiogram analysis (normal range in brackets).
Sudden cardiac death.
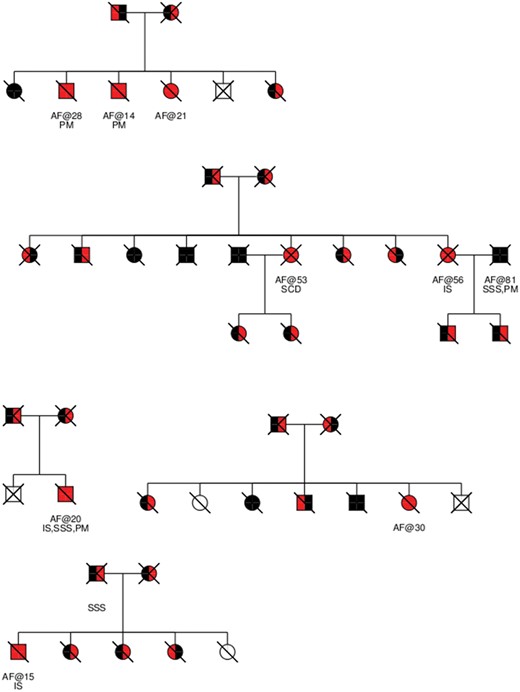
The five pedigrees containing the eight homozygous carriers of c.234delC in MYL4. Homozygous wild-type individuals are coloured black, c.234delC homozygotes are coloured red, heterozygotes who received their c.234delC allele from their father are coloured red/black, and ones who received the c.234delC allele from their mother black/red. Individuals whose genotype could not be inferred are coloured white. Deceased individuals are stricken through with a forward leaning line. Individuals who were genotyped are marked gtd. Under each individual is an indication of whether the individual had been diagnosed with atrial fibrillation and the age at onset after the ‘@’ sign. The presence of other relevant phenotypes is indicated: Sick sinus syndrome (SSS), pacemaker implantation (PM), ischemic stroke (IS) and sudden cardiac death (SCD).
We also found, through detailed medical record review, that five of the six homozygotes with EMR diagnoses of AF had documented earlier onset than the EMR indicated (Table1). All eight homozygous carriers of the MYL4 c.234delC frameshift deletion had thus been diagnosed with early-onset AF (Table1).
Three of the chip-typed homozygotes are full siblings (Figure3), diagnosed with AF at the ages of 14, 21 and 28 (Table1). Another case presented with AF during pregnancy at age 21, but reported having had palpitations since 12 years of age.
To test for association of heterozygous carriers of c.234delC with AF, we removed the eight known homozygotes and repeated the multiplicative test. Significance was neither observed with early-onset AF (OR = 1.09, 95% CI: 0.67–1.78, P = 0.73) nor AF at any age (OR = 1.22, 95% CI: 0.98–1.53, P = 0.080).
The c.234delC variant was estimated to have been introduced to Iceland about 27 generations or approximately 807 years ago.7,12,15 The variant is not present in the available 1000 Genomes data16 or the Exome Variant Server data17 and no carriers were found on genotyping c.234delC in AF sample sets from Hong Kong (258 AF cases, 2125 controls) and Norway (357 AF cases, 323 controls).
Clinical presentation of the MYL4 c.234delC deletion
Seven of the eight homozygotes had paroxysmal AF attacks following the initial diagnosis and three have since developed permanent AF. Three had pacemakers implanted between 40 and 47 years of age due to sick sinus syndrome and one suffered sudden cardiac death at the age of 64 (Table1). None of the homozygous carriers had apparent structural heart disease at the time of AF diagnosis including no evidence of left ventricular hypertrophy (LVH) or conduction abnormalities on ECGs (Table1). Echocardiograms were available for seven homozygotes and all had normal LV size and normal ejection fraction (EF) (Table2) at diagnosis. The latest available follow up studies 31–47 years after the initial diagnosis showed that two patients, both diagnosed in the interim with hypertension, had developed mild concentric LVH on echocardiograms (wall thickness 1.3–1.4 cm) with comparable voltage changes on ECG (Table2). Both also developed mild-LV dilatation and one mildly depressed EF. One additional homozygote developed mild LV dilatation and another mildly depressed EF.
Echocardiogram results for the eight homozygous carriers of c.234delC in MYL4
| Early echocardiogram | Late echocardiogram | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Current age | Age at diagnosis of AFa and early echo | IVS (cm) [0.8–1.3] | LVPW (cm) [0.8– 1.3] | EDD (cm) [3.7– 5.7] | LA (cm) [2.5–4.4] | EF (%) [55–70] | Age at latest echo | IVS (cm) [0.8–1.3] | LVPW (cm) [0.8– 1.3] | EDD (cm) [3.7– 5.7] | LA (cm) [2.5–4.4] | EF (%) [55–70] |
| Male | 47 | 14 | 1.0 | 1.0 | 5.4 | 4.4 | 60 | 49 | 1.1 | 1.2 | 5.0 | ↑b | 40 |
| Female | 53 | 21 | 0.9 | 0.9 | 5.1 | 4.2 | 55 | 55 | 1.0 | 1.0 | 6.0 | 4.7 | 55 |
| Male | 58 | 28 | 1.0 | 0.9 | 5.3 | 4.2 | 65 | 60 | 1.2 | 1.2 | 5.2 | ↑b | 55 |
| Female | Died 64 | 53 | — | — | — | — | — | — | — | — | — | — | — |
| Female | Died 58 | 56 | 1.0 | 1.0 | 5.3 | 5.5 | 55 | — | — | — | — | — | — |
| Male | 57 | 20 | 1.1 | 1.0 | 5.6 | ↑b | 65 | 51 | 1.2 | 1.0 | 5.8 | 5.6 | 65 |
| Female | 66 | 30 | Normc | Normc | Normc | – | 52d | 64 | 1.4 | 1.3 | 6.2 | 5.4 | 65 |
| Male | 59 | 15 | 1.1 | 1.1 | 5.4 | 4.3 | 62 | 62 | 1.4 | 1.4 | 6.0 | 6.1 | 45 |
| Early echocardiogram | Late echocardiogram | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Current age | Age at diagnosis of AFa and early echo | IVS (cm) [0.8–1.3] | LVPW (cm) [0.8– 1.3] | EDD (cm) [3.7– 5.7] | LA (cm) [2.5–4.4] | EF (%) [55–70] | Age at latest echo | IVS (cm) [0.8–1.3] | LVPW (cm) [0.8– 1.3] | EDD (cm) [3.7– 5.7] | LA (cm) [2.5–4.4] | EF (%) [55–70] |
| Male | 47 | 14 | 1.0 | 1.0 | 5.4 | 4.4 | 60 | 49 | 1.1 | 1.2 | 5.0 | ↑b | 40 |
| Female | 53 | 21 | 0.9 | 0.9 | 5.1 | 4.2 | 55 | 55 | 1.0 | 1.0 | 6.0 | 4.7 | 55 |
| Male | 58 | 28 | 1.0 | 0.9 | 5.3 | 4.2 | 65 | 60 | 1.2 | 1.2 | 5.2 | ↑b | 55 |
| Female | Died 64 | 53 | — | — | — | — | — | — | — | — | — | — | — |
| Female | Died 58 | 56 | 1.0 | 1.0 | 5.3 | 5.5 | 55 | — | — | — | — | — | — |
| Male | 57 | 20 | 1.1 | 1.0 | 5.6 | ↑b | 65 | 51 | 1.2 | 1.0 | 5.8 | 5.6 | 65 |
| Female | 66 | 30 | Normc | Normc | Normc | – | 52d | 64 | 1.4 | 1.3 | 6.2 | 5.4 | 65 |
| Male | 59 | 15 | 1.1 | 1.1 | 5.4 | 4.3 | 62 | 62 | 1.4 | 1.4 | 6.0 | 6.1 | 45 |
Shown are the sex and current age.
Age at diagnosis of AF according to review of medical records and the results of early echocardiogram (the earliest available echocardiogram) and late echocardiogram (the most recent available echocardiogram). The echocardiogram measurements reported are the interventricular septum thickness (IVS), left ventricular posterior wall thickness (LVPW), end diastolic dimension (EDD), left atrial size (LA) and ejection fraction (EF). Normal ranges of all measurements are given in brackets.
Indicates increased size, but numerical assessment not available.
Normal, exact numbers not available.
Derived from ventriculogram rather than echocardiogram.
Echocardiogram results for the eight homozygous carriers of c.234delC in MYL4
| Early echocardiogram | Late echocardiogram | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Current age | Age at diagnosis of AFa and early echo | IVS (cm) [0.8–1.3] | LVPW (cm) [0.8– 1.3] | EDD (cm) [3.7– 5.7] | LA (cm) [2.5–4.4] | EF (%) [55–70] | Age at latest echo | IVS (cm) [0.8–1.3] | LVPW (cm) [0.8– 1.3] | EDD (cm) [3.7– 5.7] | LA (cm) [2.5–4.4] | EF (%) [55–70] |
| Male | 47 | 14 | 1.0 | 1.0 | 5.4 | 4.4 | 60 | 49 | 1.1 | 1.2 | 5.0 | ↑b | 40 |
| Female | 53 | 21 | 0.9 | 0.9 | 5.1 | 4.2 | 55 | 55 | 1.0 | 1.0 | 6.0 | 4.7 | 55 |
| Male | 58 | 28 | 1.0 | 0.9 | 5.3 | 4.2 | 65 | 60 | 1.2 | 1.2 | 5.2 | ↑b | 55 |
| Female | Died 64 | 53 | — | — | — | — | — | — | — | — | — | — | — |
| Female | Died 58 | 56 | 1.0 | 1.0 | 5.3 | 5.5 | 55 | — | — | — | — | — | — |
| Male | 57 | 20 | 1.1 | 1.0 | 5.6 | ↑b | 65 | 51 | 1.2 | 1.0 | 5.8 | 5.6 | 65 |
| Female | 66 | 30 | Normc | Normc | Normc | – | 52d | 64 | 1.4 | 1.3 | 6.2 | 5.4 | 65 |
| Male | 59 | 15 | 1.1 | 1.1 | 5.4 | 4.3 | 62 | 62 | 1.4 | 1.4 | 6.0 | 6.1 | 45 |
| Early echocardiogram | Late echocardiogram | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sex | Current age | Age at diagnosis of AFa and early echo | IVS (cm) [0.8–1.3] | LVPW (cm) [0.8– 1.3] | EDD (cm) [3.7– 5.7] | LA (cm) [2.5–4.4] | EF (%) [55–70] | Age at latest echo | IVS (cm) [0.8–1.3] | LVPW (cm) [0.8– 1.3] | EDD (cm) [3.7– 5.7] | LA (cm) [2.5–4.4] | EF (%) [55–70] |
| Male | 47 | 14 | 1.0 | 1.0 | 5.4 | 4.4 | 60 | 49 | 1.1 | 1.2 | 5.0 | ↑b | 40 |
| Female | 53 | 21 | 0.9 | 0.9 | 5.1 | 4.2 | 55 | 55 | 1.0 | 1.0 | 6.0 | 4.7 | 55 |
| Male | 58 | 28 | 1.0 | 0.9 | 5.3 | 4.2 | 65 | 60 | 1.2 | 1.2 | 5.2 | ↑b | 55 |
| Female | Died 64 | 53 | — | — | — | — | — | — | — | — | — | — | — |
| Female | Died 58 | 56 | 1.0 | 1.0 | 5.3 | 5.5 | 55 | — | — | — | — | — | — |
| Male | 57 | 20 | 1.1 | 1.0 | 5.6 | ↑b | 65 | 51 | 1.2 | 1.0 | 5.8 | 5.6 | 65 |
| Female | 66 | 30 | Normc | Normc | Normc | – | 52d | 64 | 1.4 | 1.3 | 6.2 | 5.4 | 65 |
| Male | 59 | 15 | 1.1 | 1.1 | 5.4 | 4.3 | 62 | 62 | 1.4 | 1.4 | 6.0 | 6.1 | 45 |
Shown are the sex and current age.
Age at diagnosis of AF according to review of medical records and the results of early echocardiogram (the earliest available echocardiogram) and late echocardiogram (the most recent available echocardiogram). The echocardiogram measurements reported are the interventricular septum thickness (IVS), left ventricular posterior wall thickness (LVPW), end diastolic dimension (EDD), left atrial size (LA) and ejection fraction (EF). Normal ranges of all measurements are given in brackets.
Indicates increased size, but numerical assessment not available.
Normal, exact numbers not available.
Derived from ventriculogram rather than echocardiogram.
Three homozygotes suffered ischemic stroke 35—56 years of age (Table1). One was female but none had other known risk factors for stroke prior to the event.18 One of these individuals later developed hypertension.
Discussion
We have found a frameshift deletion in the sarcomere gene MYL4 that causes early-onset AF. All eight homozygous carriers in our study were diagnosed with early-onset AF and six were diagnosed before they were 30 years old, a very early age for this arrhythmia.
The frameshift mutation is in strong LD with several other non-coding sequence variants. It is possible that the observed association is caused by one of these other variants, but predicted loss of function variants are much more likely to associate with disease and to be the causal mutation than non-coding variants.
Myosin is a major component of the sarcomere, the building block the of the striated muscle contractile system.19 Myosin is an ATPase cellular motor protein composed of two heavy chains and two pairs of light chains: the essential (ELC) and regulatory light chains. MYL4 encodes a 197 amino acid myosin ELC, also known as atrial light chain 1, expressed in cardiac and skeletal muscle of the fetus20 and in the atria after birth.21 Ventricular expression of MYL4 is reactivated in congenital heart diseases and cardiomyopathies and has been associated with improved contractile function.22
To date, the only in vivo studies on the function of MYL4 were performed in zebrafish.23,24 Complete loss of function of the MYL4 orthologue cmlc1 in zebrafish leads to absence of sarcomere structures and loss of cardiac contractility. Interestingly, a zebrafish mutant carrying a C-terminal truncating mutation in cmlc1 shows severely reduced cardiac contractility but sarcomere structures appear normal, leading the authors to suggest a regulatory as well as a structural function for MYL424,24
Sequence variants in MYL4 have not been associated with arrhythmias or other diseases previously. Arrhythmias are, however, commonly seen in cardiomyopathies caused by mutations in ventricular sarcomere genes and structural myocardial disease is a well-established major risk factor for conduction disturbance. For example, AF is found in over 20% of patients with hypertrophic cardiomyopathy,25 a disease caused by mutations in genes encoding proteins of and associated with the sarcomere, including MYL3, the ventricular equivalent of MYL4.21 In those cases, AF may be a consequence of structural and hemodynamic changes in the atria due to left ventricular disease.
There was no convincing evidence of overt structural heart disease associated with the MYL4 deletion in our study. However, mild or subclinical myopathy of the atria cannot be excluded based on the available data. During follow up, four patients developed mild ventricular cardiomyopathies, possibly the consequence of longstanding AF or other risk factors including hypertension. We have previously described the absence of apparent cardiomyopathy from carriers of a missense mutation in another atrial sarcomere gene, MYH6, that is associated with sick sinus syndrome and AF.7
Another similarity of the MYL4 deletion with the MYH6 sarcomere mutation is the association with sick sinus syndrome and pacemaker implantation. Three of the eight homozygous carriers had required pacemaker implantation due to sick sinus syndrome, that while commonly seen with AF, is an uncommon syndrome in the young and middle aged population.26 For comparison, of the 1944 individuals in our dataset who were diagnosed with early-onset AF (irrespective of genotype information), 128 (6.6%) have had a pacemaker implantation at any age.
Interestingly, three of the eight individuals suffered ischemic stroke before the age of 60 years. Two would have been classified as low risk by the CHA2DS2-VASc criteria and one as low-moderate risk, the risk factor in that case being female gender. However gender is not considered a strong risk factor for stroke under the age of 65 years.27 This suggests an unusually high risk of stroke in these patients, potentially related to underlying atrial cardiomyopathy, and raises the possibility of genotype-based risk stratification of atrial thrombus formation.
In conclusion, mutations in atrial sarcomere genes causing atrial arrhythmias suggest an important contribution of the cardiac contractile system to cardiac conduction and AF even in the absence of apparent cardiomyopathy. These findings may provide novel mechanistic insight into the pathophysiology of this complex arrhythmia.
Supplementary material
Supplementary material is available at European Heart Journal online.
Conflict of interest: none declared.
References
Exome Variant Server NGESPE, Seattle, WA. http://evs.gs.washington.edu/EVS/ (30 July 2015).
Author notes
See page 35 for the editorial comment on this article (doi: )


{kind=link}
{kind=link}